Winnowmap is a long-read mapping algorithm optimized for mapping ONT and PacBio reads to repetitive reference sequences. Winnowmap development began on top of minimap2 codebase, and since then we have incorporated the following two ideas to improve mapping accuracy within repeats.
-
Winnowmap implements a novel weighted minimizer sampling algorithm (>=v1.0). This optimization was motivated by the need to avoid masking of frequently occurring k-mers during the seeding stage in an efficient manner, and achieve better mapping accuracy in complex repeats (e.g., long tandem repeats) of the human genome. Using weighted minimizers, Winnowmap down-weights frequently occurring k-mers, thus reducing their chance of getting selected as minimizers. Users can refer to this paper for more details. This idea is helpful to preserve the theoretical guarantee of minimizer sampling technique, i.e., if two sequences share a substring of a specified length, then they must be guaranteed to have a matching minimizer.
-
We noticed that the highest scoring alignment doesn't necessarily correspond to correct placement of reads in repetitive regions of T2T human chromosomes. In the presence of a non-reference allele within a repeat, a read sampled from that region could be mapped to an incorrect repeat copy because the standard pairwise sequence alignment scoring system penalizes true variants. This is also sometimes referred to as allelic bias. To address this bias, we introduced and implemented an idea of using minimal confidently alignable substrings (>=v2.0). These are minimal-length substrings in a read that align end-to-end to a reference with mapping quality score above a user-specified threshold. This approach treats each read mapping as a collection of confident sub-alignments, which is more tolerant of structural variation and more sensitive to paralog-specific variants (PSVs). Our most recent paper desribes this concept and benchmarking results.
Clone source code from master branch or download the latest release.
git clone https://github.com/marbl/Winnowmap.gitWinnowmap compilation requires C++ compiler with c++11 and openmp, which are available by default in GCC >= 4.8.
cd Winnowmap
make -j8Expect winnowmap and meryl executables in bin folder.
For either mapping long reads or computing whole-genome alignments, Winnowmap requires pre-computing high frequency k-mers (e.g., top 0.02% most frequent) in a reference. Winnowmap uses meryl k-mer counting tool for this purpose.
- Mapping ONT or PacBio-hifi WGS reads
meryl count k=15 output merylDB ref.fa
meryl print greater-than distinct=0.9998 merylDB > repetitive_k15.txt
winnowmap -W repetitive_k15.txt -ax map-ont ref.fa ont.fq.gz > output.sam [OR]
winnowmap -W repetitive_k15.txt -ax map-pb ref.fa hifi.fq.gz > output.sam- Mapping genome assemblies
meryl count k=19 output merylDB asm1.fa
meryl print greater-than distinct=0.9998 merylDB > repetitive_k19.txt
winnowmap -W repetitive_k19.txt -ax asm20 asm1.fa asm2.fa > output.samFor the genome-to-genome use case, it may be useful to visualize the dot plot. This perl script can be used to generate a dot plot from paf-formatted output. In both usage cases, pre-computing repetitive k-mers using meryl is quite fast, e.g., it typically takes 2-3 minutes for the human genome reference.
When comparing Winnowmap (v1.0) to minimap2 (v2.17-r954), we observed a reduction in the mapping error-rate from 0.14% to 0.06% in the recently finished human X chromosome, and from 3.6% to 0% within the highly repetitive X centromere (3.1 Mbp). Winnowmap improves mapping accuracy within repeats and achieves these results with sparser sampling, leading to better index compression and competitive runtimes. By avoiding masking, we show that Winnowmap maintains uniform minimizer density.
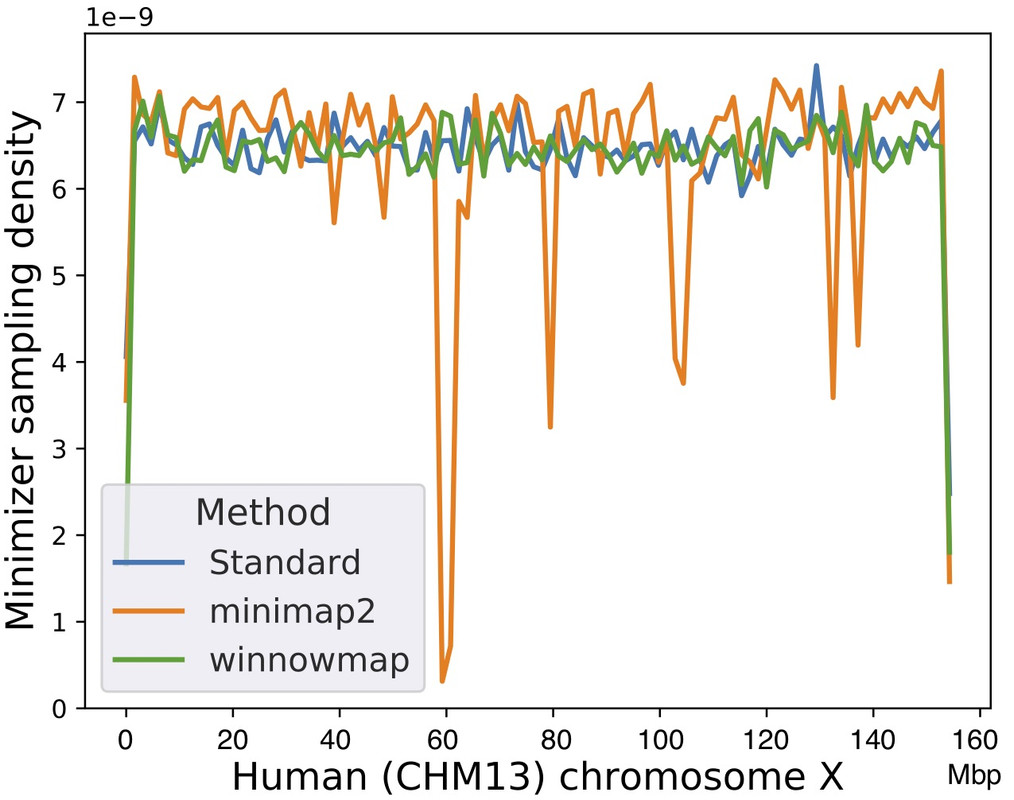
Minimizer sampling density using a human X chromosome as the reference, with the centromere positioned between 58 Mbp and 61 Mbp. ‘Standard’ method refers to the classic minimizer sampling algorithm from Roberts et al., without any masking or modification.
- Chirag Jain, Arang Rhie, Nancy Hansen, Sergey Koren and Adam Phillippy. "Long-read mapping to repetitive reference sequences using Winnowmap2". Nature Methods, 2022.
- Chirag Jain, Arang Rhie, Haowen Zhang, Chaudia Chu, Brian Walenz, Sergey Koren and Adam Phillippy. "Weighted minimizer sampling improves long read mapping". Bioinformatics (ISMB proceedings), 2020.