Learning objectives
- Clean up (de contaminate) the assembly
- Assign chromosome using an existing reference genome
- Detect remaining errors with traditional linear-reference mapping
- Understand spectra-cn plots
The proper way is to identify mitochondrial genoomes (MT), chloroplast, or any other foreign contamination.
The Vertebrate Genomes Project has been using Mash screen and Mash distance for this purpose.
Recently, NCBI released their contamination detection tool FCS runnable locally. They highly recommend to run this tool before submitting any assembled genomes.
Additionally, most of the additional small sequences comes from the excessive copies of rDNA, that could not be resolved in Verkko.
The latest version of Verkko v1.3.1 comes with a new feature, --screen human, which tries to auto-detect rDNA, MT, and Epstein-Barr Virus (EBV) sequences, and builds a circularized consensus. Other contaminants can be filtered using --screen <label> </path/to/single/sequence.fasta>. All contigs matching a contaminant are removed from assembly.fasta and placed in their own file. The exemplar contig is circularized and saved separately.
For simplicity, let’s remove contigs <100kb.
cd ~/day3_assembly_evaluation/Col-0/asm/
# Download the final assembly.fasta if Verkko was not run in the previous step
wget -c https://s3-us-west-2.amazonaws.com/human-pangenomics/T2T/scratch/a_thal/Col-0/asm/assembly.fasta
# Let's do the renaming in a separate dir as it could become messy
cd ~/day3_assembly_evaluation/Col-0/rename
ln -s ../asm/assembly.fasta
# Indexing to enable random access
samtools faidx assembly.fasta
# Collect fasta sequence IDs > 100 kb
cat assembly.fasta.fai | awk '$2>100000 {print $1}' > origin.list
# Extract from origin.list
samtools faidx -r origin.list assembly.fasta > assembly.100k.fastaIf there is a known reference, it's often convenient to assign chromosome names to contigs prior of doing any further analysis.
This will also help determining where the contigs are broken, and perhaps could indicate if additional scaffolding would be beneficial.
In this course, we will use the latest A. thaliana reference genome (TAIR10.1). I have renamed the RefSeq assembly sequence IDs for our convenience. It is available at /opt/assembly_data/TAIR10.1.fasta or downloadable from aws.
Here we are using MashMap v2.0 to quickly obtain an approximate alignment. This step could be replaced with wfmash or the latest MashMap v3.0, but may require some additional tunings due to output format differences.
cd ~/day3_assembly_evaluation/Col-0/rename
ln -s /opt/assembly_data/TAIR10.1.fasta
mashmap \
-r TAIR10.1.fasta \
-q assembly.100k.fasta \
-f one-to-one --pi 95 -s 100000 \
-t 16 -o mashmap/mashmap.outWe have learnt about wfmash in the previous course. The --pi P and -s S are features of MashMap, which lets you tune to report mappings with identity > P on segment size of S.
Note we are using a relatively high --p as we are comparing assemblies from the same species.
There are cases where no reference species is available. In such cases, we still would like to orient to the closest species is available. Depending on how close the reference species is, consider lowering --p parameters, perhaps --p 85, if that does not work, additionally lower the segment size, to something like -s 50000.
generateDotPlot png large mashmap/mashmap.outDownload out.png and see how the dotplot looks like.
What is your take on this?
Answer:
We can see that our new assembly is near T2T (especially chr5 has no gaps!), assembling most centromeric sequences of all the 5 chromosomes, albeit we see some breaks in chr1 (3 pieces), chr2 (3 pieces), and chr4 (3 pieces).Take a look at mashmap/mashmap.out to see the orientation of each contigs.
As this is a very simple genome, we will do some manual works to re-orient and ordering them.
awk '{print $1"\t"$6"\t"$5}' mashmap/mashmap.out | sort -uBelow is a simple view of the contigs and their relative orientation to the reference.
``` contig-0000075 chr5 - contig-0000088 chr1 + contig-0000089 chr2 - contig-0000104 chr3 - contig-0000104 chr3 + contig-0000138 chr1 + contig-0000182 chr2 + contig-0000357 chrCP - contig-0000392 chr4 - contig-0000403 chr1 + contig-0000531 chr2 - contig-0000572 chr2 + contig-0000651 chr4 - ```Note that we have one contig in both + and - directions.
contig-0000104 26150228 9600000 10099999 - chr3 23459830 13732848 14196395 99.3596
contig-0000104 26150228 0 9399999 - chr3 23459830 14142835 23456936 99.8913
contig-0000104 26150228 10200000 10299999 + chr3 23459830 13676292 13776291 99.1654
contig-0000104 26150228 10300000 10399999 - chr3 23459830 13584419 13684418 99.4562
contig-0000104 26150228 12500000 26150227 - chr3 23459830 0 13643278 99.9832
This indicates a possible inversion error in the TAIR10.1 reference genome, around chr3:23459830-14142835, as they come from the same species, or likely a rearrangement that happened in the sequenced sample.
As we want to assign the contig to match the majority of the orientation to match the reference, let's ignore the + alignment and assign contig-0000104 to the - orientation.
Feel free to use your own way of doing this, or just simply follow the steps below.
# Collect sequences to reverse-complement (rc)
awk '{print $1"\t"$6"\t"$5}' mashmap/mashmap.out | sort -u | awk '$NF=="-" {print $1}' > rc.list
samtools faidx -r rc.list assembly.100k.fasta | seqtk seq -r - > assembly.100k.rc.fasta
# Sequences we need to remain in its original orientation, except contig-0000104
awk '{print $1"\t"$6"\t"$5}' mashmap/mashmap.out | sort -u | awk '$NF=="+" {print $1}' | grep -v contig-0000104 > ps.list
samtools faidx -r ps.list assembly.100k.fasta > assembly.100k.ps.fasta
# The most under-appreciated assembler cat
cat assembly.100k.ps.fasta assembly.100k.rc.fasta > assembly.100k.reorient.fasta
samtools faidx assembly.100k.reorient.fasta
# Now we want to re-name the contig names to chrN
cat assembly.100k.reorient.fasta.fai | awk '{print $1}' | sort -k1 > oriented.list
cat mashmap/mashmap.out | awk '{print $1"\t"$6}' | sort -u > rename.tmp.map
Let's take a break and check again how we want to order the chromosomal pieces.
Edit rename.tmp.map and save it as rename.map to match the order in TAIR10.1. Use the out.png, or directly the mashmap/mashmap.out to order the contigs.
_This step could be replaced with the following, which simply orders the duplicated entries with an additional `_2`,`_3`, ... ._
```shell awk '{ chrCount[$2]++; if (chrCount[$2] > 1) { print $1, $2"_"chrCount[$2] } else { print $1, $2 } }' rename.tmp.map > rename.map ```Let's see how the final rename.map looks like.
cat rename.map | sort -k2 # Check how the output looks like, in reference ordercontig-0000403 chr1_1
contig-0000088 chr1_2
contig-0000138 chr1_3
contig-0000182 chr2_1
contig-0000572 chr2_2
contig-0000089 chr2_3
contig-0000531 chr2_4
contig-0000104 chr3
contig-0000392 chr4_1
contig-0000651 chr4_2
contig-0000075 chr5
contig-0000357 chrCP
This is my hacky way of replacing fasta sequence name entries since 2019.
cat rename.map | sort -k2 | awk '{print $1}' > origin.list
cat rename.map | sort -k2 | awk '{print $2}' > rename.list
java -jar -Xmx256m $T2T_Polish/paf_util/fastaExtractFromList.jar \
assembly.100k.reorient.fasta origin.list Col0.fasta rename.listThe fastaExtractFromList.jar takes the input fasta assembly.100k.fasta, replaces the sequence entries seen in origin.list to rename.list, and outputs as Col0.fasta. It's not as fancy and does not re-order the scaffolds in the fasta sequence. There are other tools that better support this, or you could probably write your own script.
Now as we are done with chromosome assignment, let's check again if the renamed chromosomes appear as we wanted in MashMap alignment. This time, we will do this in the mashmap dir to avoid naming collision in generateDotPlot.
mkdir -p mashmap_Col0_to_TAIR && cd mashmap_Col0_to_TAIR
mashmap -r ../TAIR10.1.fasta -q ../Col0.fasta -f one-to-one --pi 95 -s 100000 -t 16 -o Col0_to_TAIR.out
generateDotPlot png large Col0_to_TAIR.out
mv out.png ../Col0_to_TAIR.png
cd ..Open `Col0_to_TARI.png`. Check the chromosome pieces are in order.
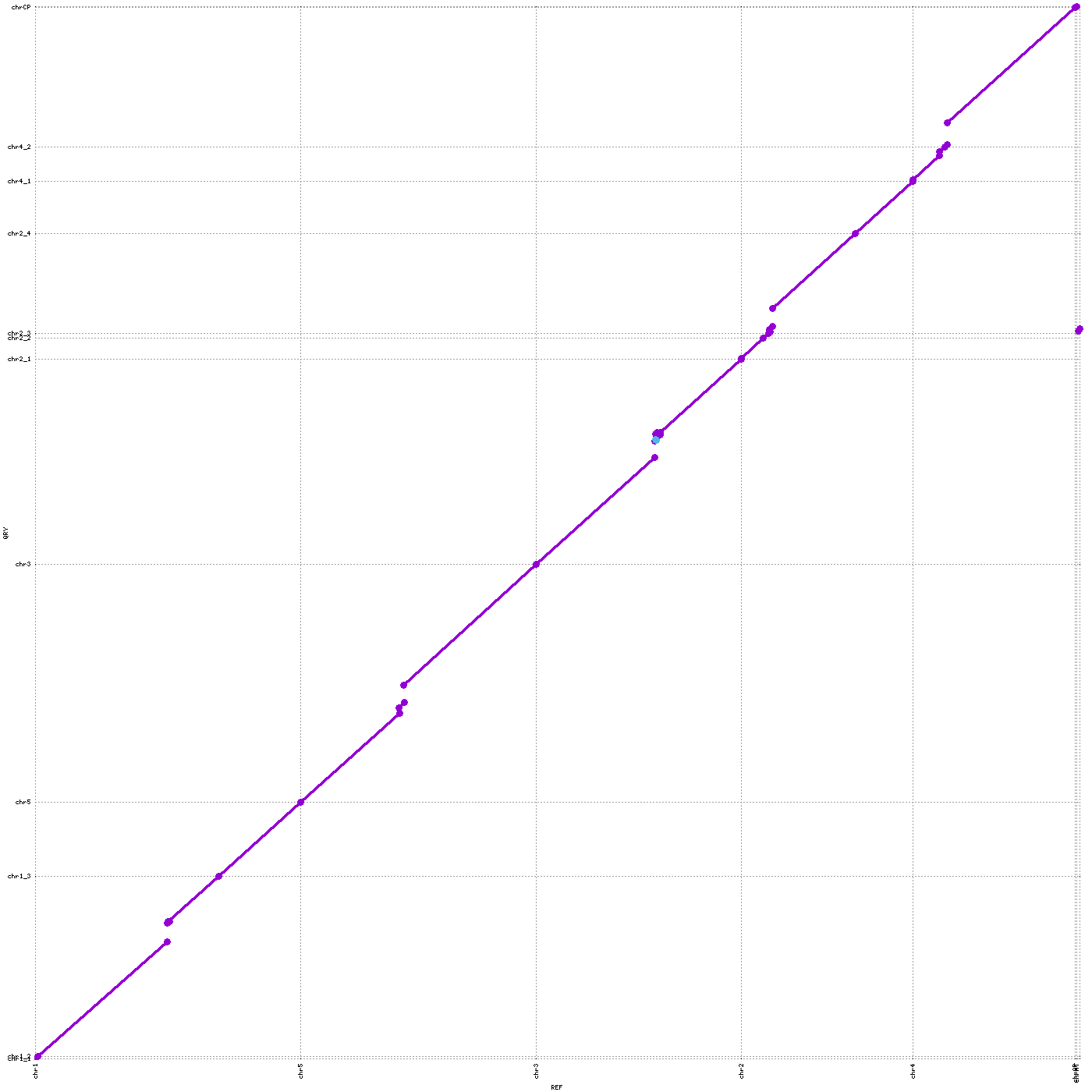The first question you'd ask, given the high continuity, would be "how many chromosomes are T2T"?
It is surprisingly not so easy in some non-human genomes to get the genomes T2T.
Let's see if A. thaliana is one of those.
The plant telomere motifs, including A. thaliana, are known to be TTTAGGG.
Heng Li recently added seqtk telo to make our jobs easier.
seqtk telo -m TTTAGGG Col0.fasta > Col0.teloSadly, there are no telomere motifs found. There might be some other complex sub-telomeric microsatellite repeats, and/or we simply do not have enough ONT UL reads to resolve the telomeric ends.
At home: Try step 2-3 again on other A. thaliana genomes. Why is it so difficult to get T2T?
An assembly is an hypothesis, and we want to test if the assembly is concordant with the reads.
We will align the HiFi and ONT data back to the assembly and see if there are any coverage abnormalities.
Some are associated with sequencing biases (2-mer microsatellite repeats, i.e. GAs or TAs), so we'd like to annotate if any of the low or high coverage region are associated with these.
Consensus base errors are also often associated with to low HiFi coverage.
Therefore, we will prepare some files first
- Winnowmap primary alignments (pre-ran bam files are available under ~/day3_assembly_evaluation/Col-0/mapping)
- Seqrequester microsatellite sequence pattern tracks
- Merqury error kmer tracks
The Winnowmap alignment has been pre-computed, for the sake of time.
cd ~/day3_assembly_evaluation/Col-0/mapping/
ln -s ../rename/Col0.fasta
cd hifi/
# Align HiFi and ONT reads ← skip this part for now as we don’t have enough time :)
echo /opt/assembly_data/Col-0.hifi.q20.50x.fq.gz > input.fofn
$T2T_Polish/winnowmap/init.sh ../Col0.fasta
$T2T_Polish/winnowmap/map.sh ../Col0.fasta map-pb
$T2T_Polish/winnowmap/merge.sh Col0.hifi
$T2T_Polish/winnowmap/filt.sh Col0.hifi.bam
## skip end
# Do this for HiFi and ONT bams - the $tools/sam2paf.sh requires k8; which is not installed. We are skipping this step.
# $T2T_Polish/coverage/sam2paf.sh Col0.hifi.pri.bam Col0.hifi.pri.paf
# Download the .paf files from aws:
wget https://s3-us-west-2.amazonaws.com/human-pangenomics/T2T/scratch/a_thal/Col-0/mapping/hifi/Col0.hifi.pri.paf
# Also for ONT:
cd ../ont/
wget https://s3-us-west-2.amazonaws.com/human-pangenomics/T2T/scratch/a_thal/Col-0/mapping/ont/Col0.ont.pri.paf
# Go back and finish this step for both hifi and ont
cd ../hifi/
$T2T_Polish/coverage/init.sh Col0.fasta Col0Let's collect the 2-mer microsatellites with seqrequester.
# Profile 2-mer microsatellite sequence patterns
cd ~/day3_assembly_evaluation/Col-0/pattern
ln -s ../rename/Col0.fasta
ln -s ../rename/Col0.fasta.fai
$T2T_Polish/pattern/microsatellites.sh Col0.fasta
# OUTPUT: Col0/Col0.microsatellite.*.128.wig
# or Col0/Col0.microsatellite.*.128.bw if ucsc tools is installedLet's run Merqury.
cd ~/day3_assembly_evaluation/Col-0/merqury
ln -s ../meryl/Col-0.illumina.k21.meryl
$MERQURY/merqury.sh Col-0.illumina.k21.meryl ../rename/Col0.fasta Col-0
bedtools merge -i Col0_only.bed > Col0_only.mrg.bedWhat's the QV?
cat Col-0.qvWe are going back to the mapping directory.
# symlink all files needed in mapping/
cd ~/day3_assembly_evaluation/Col-0/mapping/
ln -s ../merqury/Col0_only.mrg.bed Col0.error.bed
ln -s ../pattern
touch Col0.exclude.bed # usually, this becomes the rDNA region or gaps
# symlink each files again under hifi/ and ont/
# Required: pattern/ver/, ver.bed, ver.error.bed, ver.exclude.bed, ver.telo.bed
ln -s ../Col0.bed
ln -s ../Col0.exclude.bed
ln -s ../Col0.telo.bed
ln -s ../Col0.error.bed
ln -s ../patternThe issues.sh is generating a lot of .wig files, so we are generating them separately for each platform.
Feel free to poke around to see what each files represent.
cd hifi/
$T2T_Polish/coverage/issues.sh Col0.hifi.pri.paf "HiFi" Col0 HiFi
# Do the same on ONT paf
cd ../ont/
$T2T_Polish/coverage/issues.sh Col0.ont.pri.paf "ONT" Col0 ONT
Now, load everything on IGV. The essential files we need are on aws.
- The reference: Col0.fasta. Go to "Genome -> Load from URL"
Loead the tracs from URL.
mapping/hifi/*.pri.cov.wigandmapping/ont/*.pri.cov.wigmapping/hifi/*.issues.bedandmapping/ont/*.issues.bedpattern/Col-0/Col0.microsatellite.*.128.bwmapping/Col-0.error.bed