Releases: arq5x/bedtools2
Releases · arq5x/bedtools2
bedtools version 2.25.0
bedtools version 2.24.0
- The
coveragetool now takes advantage of pre-sorted intervals via the-sortedoption. This allows thecoveragetool to be much faster,
use far less memory, and report coverage for intervals in their original order in the input file. - We have changed the behavior of the
coveragetool such that it is consistent with the other tools. Specifically, coverage is now
computed for the intervals in the A file based on the overlaps with the B file, rather than vice versa. - The
subtracttool now supports pre-sorted data via the-sortedoption and is therefore much faster and scalable. - The
-nonamecheckoption provides greater tolerance for chromosome labeling when using the-sortedoption. - Support for multiple SVLEN tags in VCF format, and fixed a bug that failed to process SVLEN tags coming at the end of a VCF INFO field.
- Support for reverse complementing IUPAC codes in the
getfastatool. - Provided greater flexibility for "BED+" files, where the first 3 columns are chrom, start, and end, and the remaining columns are free-form.
- We now detect stale FAI files and recreate an index thanks to a fix from @gtamazian.
- New feature from Pierre Lindenbaum allowing the
sorttool to sort files based on the chromosome order in afaidxfile. - Eliminated multiple compilation warnings thanks to John Marshall.
- Fixed bug in handling INS variants in VCF files.
bedtools version 2.23.0
New features.
- Added
-koption to theclosesttool to report the k-closest features in one or more-bfiles. - Added
-fdoption to theclosesttool to for the reporting of downstream features in one or more-bfiles. Requires-Dto dictate how "downstream" should be defined. - Added
-fuoption to theclosesttool to for the reporting of downstream features in one or more-bfiles. Requires-Dto dictate how "downstream" should be defined. - @lindenb added a new
splittool that will split an input file into multiple sub files. Unlike UNIXsplit, it can balance the chunking of the sub files not just by number of lines, but also by total number of base pairs in each sub file. - Added a new
spacingtool that reports the distances between features in a file. - @jayhesselberth added a
-reverseoption to themakewindowstool that reverses the order of the assigned window numbers.
Bug fixes.
bedtools version 2.22.1
- When using
-sortedwithintersect,map, andclosest, bedtools can now detect and warn you when your input datasets employ different chromosome sorting orders. - Fixed multiple bugs in the new, faster
closesttool. Specifically, the -iu, -id, and -D options were not behaving properly with the new "sweeping" algorithm that was implemented for the 2.22.0 release. Many thanks to Sol Katzman for reporting these issues and for providing a detailed analysis and example files. - We FINALLY wrote proper documentation for the
closesttool.
http://bedtools.readthedocs.org/en/latest/content/tools/closest.html - Fixed bug in the
tagtool when using -intervals, -names, or -scores. Thanks to Yarden Katz for reporting this. - Fixed issues with chromosome boundaries in the
sloptool when using negative distances. Thanks to @acdaugherty! - Multiple improvements to the
fishertool. Added a-moption to thefishertool to merge overlapping intervals prior to comparing overlaps between two input files. Thanks to @brentp - Fixed a bug in
makewindowstool requiring the use of-bwith-s. - Fixed a bug in
intersectthat prevented-splitfrom detecting complete overlaps with-f 1. Thanks to @tleonardi . - Restored the default decimal precision to the
groupbytool. - Added the -prec option to the
mergeandmaptools to specific the decimal precision of the output.
bedtools version 2.22.0
Enhancements
- Multiple database support for the closest tool. The closest tool now requires sorted input, but it is between 10 and 60 times faster depending on the use case.
As an example:
➜ cat mq1.bed
chr1 80 100 q1 1 +
➜ cat mdb1.bed
chr1 5 15 d1.1 1 +
chr1 20 60 d1.2 2 -
chr1 200 220 d1.3 3 -
➜ cat mdb2.bed
chr1 15 35 db2.1 1 -
chr1 120 170 db2.2 2 -
chr1 210 230 db3 3 +
➜ cat mdb3.bed
chr1 70 90 d3.1 3 -
Find the closest interval in each B file.
➜ bedtools closest -a mq1.bed \
-b mdb1.bed mdb2.bed mdb3.bed \
-names foo bar biz
chr1 80 100 q1 1 + foo chr1 20 60 d1.2 2 -
chr1 80 100 q1 1 + bar chr1 120 170 db2.2 2 -
chr1 80 100 q1 1 + biz chr1 70 90 d3.1 3 -
Find the closest interval among all B files.
➜ bedtools closest -a mq1.bed \
-b mdb1.bed mdb2.bed mdb3.bed \
-names foo bar biz \
-mdb all
chr1 80 100 q1 1 + biz chr1 70 90 d3.1 3 -
- Support for IMPRECISE SVs in VCF format.
- Added the
-precoption to grouby to allow control over the reported decimal precision
Bug fixes
- Fixed a bug with zero length records.
- Fixed a precision bug in the
fishertool. Thanks to @brentp - Fixed a bug in the bamtofastq tool. Thanks to @ryan-williams
bedtools version 2.21.0
Version 2.21.0 (18-Sep-2014)
- Added ability to intersect against multiple -b files in the intersect tool.
- Fixed a bug causing slowdowns in the -sorted option when using -split with very large split alignments.
- Added a new fisher tool to report a P-value associated with the significance of the overlaps between two interval sets. Thanks to @brentp!
- Added a “genome” file for GRCh38. Thanks @martijnvermaat!
- Fixed a bug in the -pct option of the slop tool. Thanks to @brentp!
- Tweak to the Makefile to accomodate Intel compilers. Thanks to @jmarshall.
- Many updates to the docs from the community. Thank you!
bedtools version 2.20.1
- Fixed a float rounding bug causing occassional off-by-one issues in the slop added by the
sloptool. Thanks to @slw287r. - Fixed a bug injected in 2.19 arising when files have a single line not ending in a newline. Thanks to @cwarden45.
bedtools version 2.20.0
- The
mergetool now supports BAM input.- The
-n,-nms, and-scoresoptions are deprecated in favor of the new, substantially more flexible,-cand-ooptions. See the docs: http://bedtools.readthedocs.org/en/latest/content/tools/merge.html - It now supports the
-headeroption. - It now supports the
-Soption.
- The
- The
maptool now supports BAM input. - The
jaccardtool is now ~3 times faster.- It now supports the
-splitoption. - It now supports the
-soption. - It now supports the
-Soption.
- It now supports the
- We have fixed several CLANG compiler issues.Thanks to John Marshall for the thorough report.
- We added support for “X” and “=” CIGAR operators. Thanks to Pierre Lindenbaum.
- Fixed bugs for empty files.
- Improved the
-incloption in the shuffle tool such that the distibution is much more random. - Fixed a bug in slop when very large slop values are used.
bedtools version 2.19.1
- Bug fix to intersect causing BAM footers to be erroneously written when -b is BAM
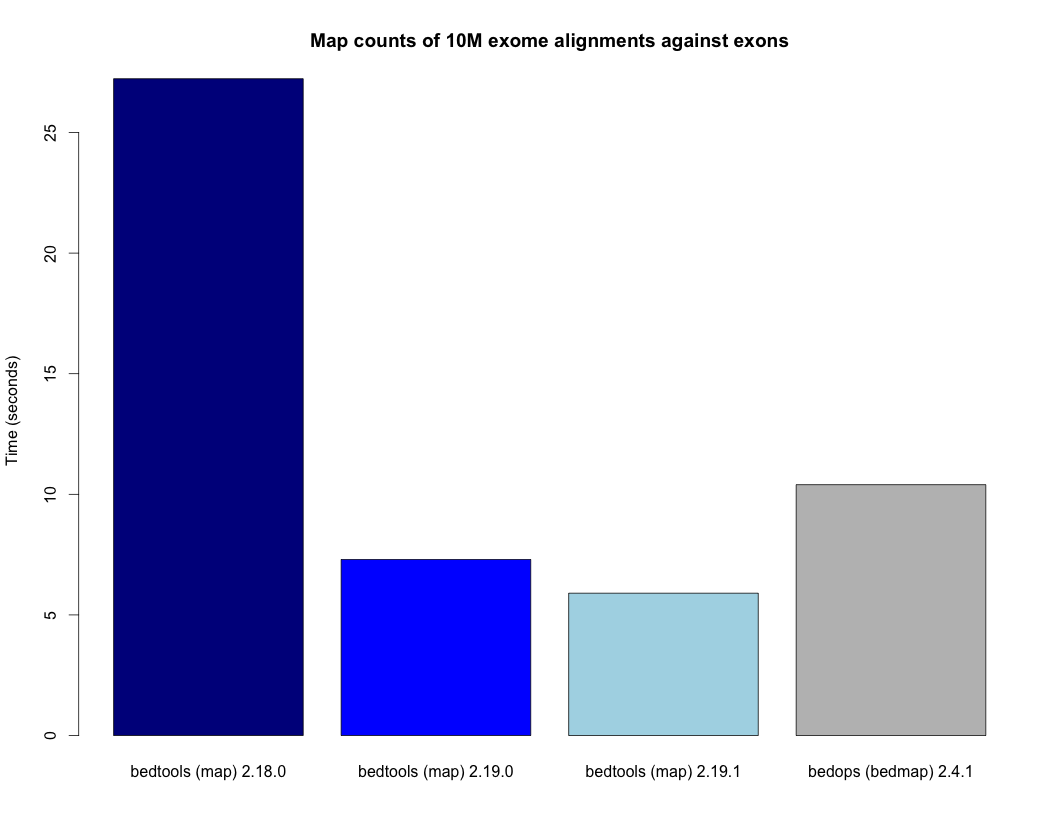
- Speedup for the map tool.
http://bedtools.readthedocs.org/en/latest/_images/map-speed-comparo.png - Map tool now allows multiple columns and operations in a single run.
http://bedtools.readthedocs.org/en/latest/content/tools/map.html#multiple-operations-and-columns-at-the-same-time
{kind=link}
bedtools version 2.19.0
v2.19.0 Setting Release-Version v2.19.0