Note : Please don't use it if you are not the fan of our biotrainee, Thanks.
GEO数据库挖掘系列知识分享课程,于2016年首发于生信菜鸟团博客:http://www.bio-info-trainee.com/2087.html
配套教学视频在B站:https://www.bilibili.com/video/av26731585/
早期目录如下:
- 第一讲:GEO,表达芯片与R
- 第二讲:从GEO下载数据得到表达量矩阵
- 第三讲:对表达量矩阵用GSEA软件做分析
- 第四讲:根据分组信息做差异分析
- 第五讲:对差异基因结果做GO/KEGG超几何分布检验富集分析
- 第六讲:指定基因分组boxplot指定基因list画热图
- 第七讲:根据差异基因list获取string数据库的PPI网络数据
- 第八讲:PPI网络数据用R或者cytoscape画网络图
- 第九讲:网络图的子网络获取
- 第十讲:hug genes如何找
2018是一个特殊的年份,我们生信技能树落地了很多线下分享会,作为创始人我身先士卒准备了非常多的干货教程,在B站免费分享 74小时的生物信息学系列视频课程: 1.3个学生的linux视频学习笔记
3.一万人陪你学习GEO数据库挖掘知识(公益视频听课笔4.记分享)
本项目主要是GEO数据库挖掘课程的代码仓库,记住,下面的每个文件夹都值得你拿出50个小时以上的时间来学习。
主要项目代码文件夹:
- GSE42872_main,主要演示GEO数据库挖掘的标准6步骤
- GSE11121_surivival,主要演示基于基因表达量分组的批量生存分析
六个任务项目文件夹:
-
task1-check-specific-genes , 主要演示检查自己感兴趣的基因在多个GSE数据集表现情况
-
task2-lncRNA-mRNA-network , 主要演示如何构建网络
-
task3-multiple-groups , 主要演示,如果样本分组信息非常复杂该如何进行下游分析
-
task4-NPC, 主要演示GEO数据库挖掘的标准6步骤,跟GSE42872_main类似
-
task5-dynamic-network-biomarker,主要演示新的算法,超脱于GSE42872_main展示的GEO数据库挖掘的标准6步骤
-
task6-GEO-TCGA,主要演示如何结合TCGA数据库
两个额外补充项目文件夹:
- airway_RNAseq,主要演示如果是RNA-seq得到的表达矩阵该如何分析,与传统芯片表达矩阵的异同
- breast_cancer_meta-analysis , 主要演示如何进行meta分析。
下面是主要代码文件夹 GSE42872_main 代码的展示:
数据是灵魂!
It always not very easy to download data if you are in China, so I also upload the file GSE42872_raw_exprSet.Rdata , you can load it directly.
if(F){
library(GEOquery)
gset <- getGEO('GSE42872', destdir=".",
AnnotGPL = F,
getGPL = F)
save(gset,'GSE42872.gset.Rdata')
}
load('GSE42872_eSet.Rdata')
b = eSet[[1]] # 看清楚,某些GSE数据集有多个平台,注意挑选
raw_exprSet=exprs(b)
group_list=c(rep('control',3),rep('case',3))
save(raw_exprSet,group_list,
file='GSE42872_raw_exprSet.Rdata')高质量的数据是保障!
Try to understand my codes, how did I filter the probes by the annotation of each microarry, and how I check the group information for the different samples in each experiment.
Including PCA and Cluster figures, as below:
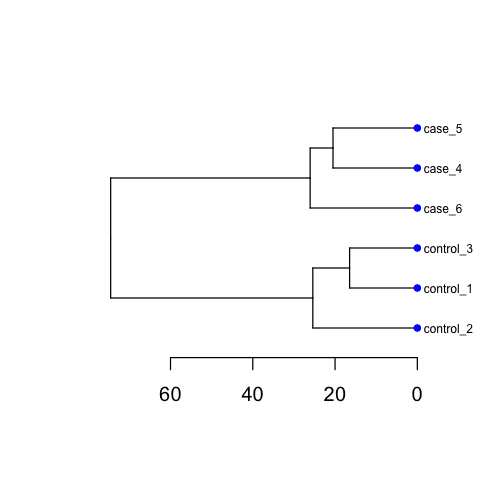
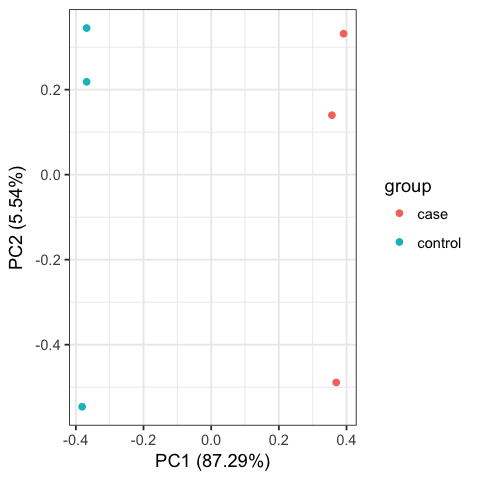
Please ensure that you do run those codes by yourself !!!
差异分析是核心流程
Normally we will do differential expression analysis for the microarray, and LIMMA is one of the best method, so I just use it. If the expression matrix(raw counts ) comes from mRNA-seq, you can also choose DESeq based on negative binomial (NB) distributions or baySeq and EBSeq.
Once DEG finished, we can choose top N genes for heatmap as below:
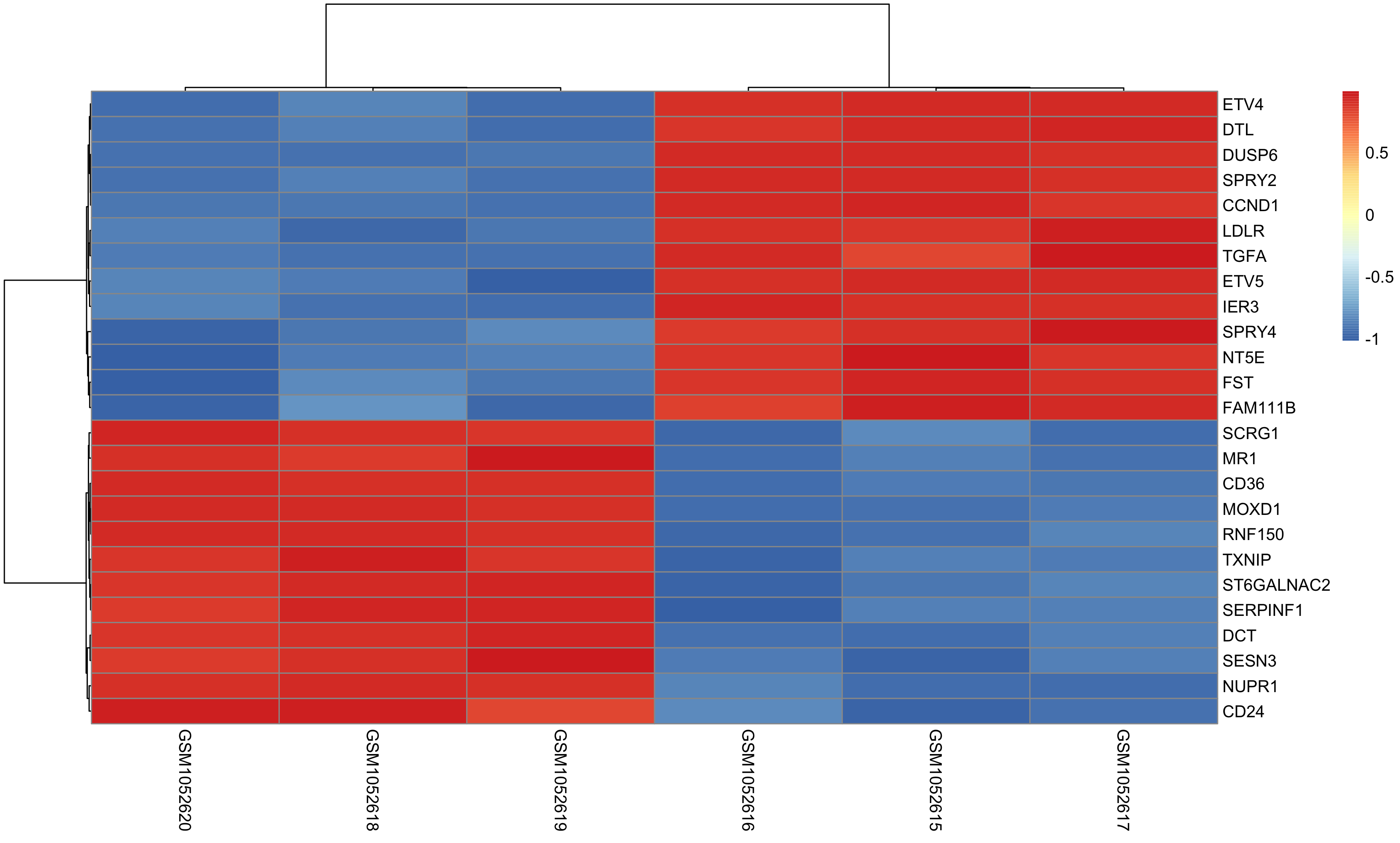
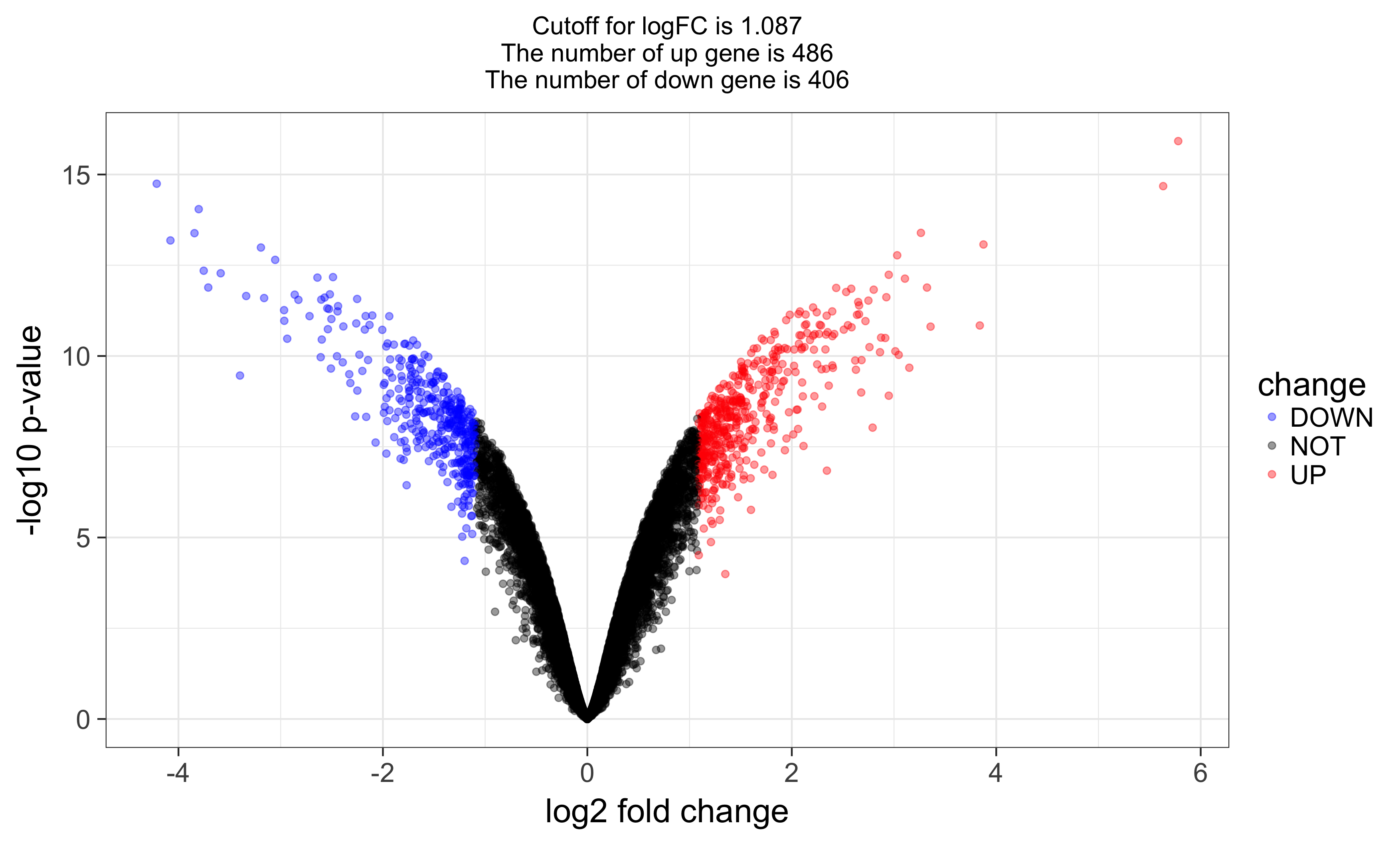
and volcano plot as below:
数据库的注释是升华!
Annotation for the significantly changed genes, over-representation test or GSEA for GO/KEGG/biocarta/rectome/MsigDB and so on.
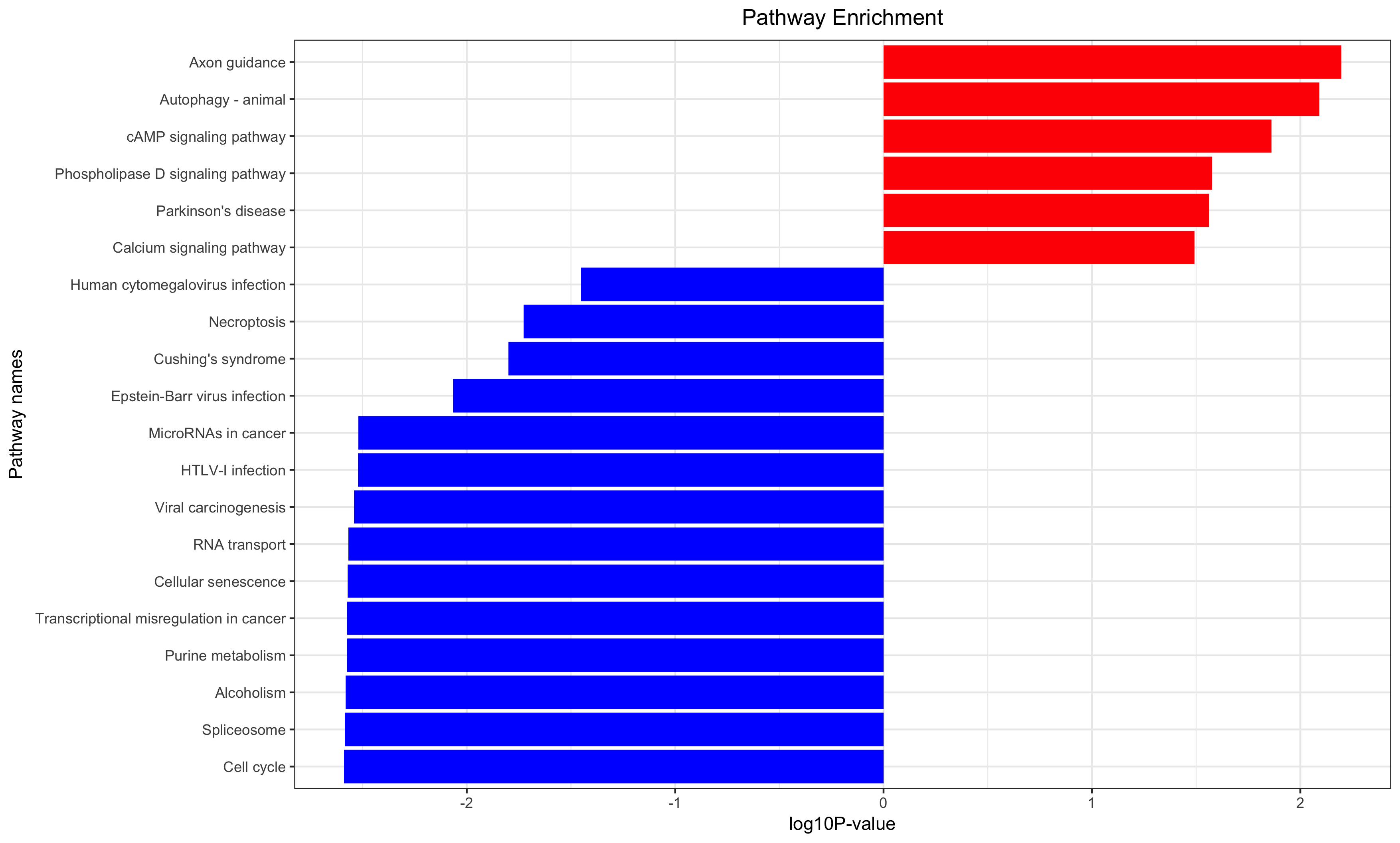
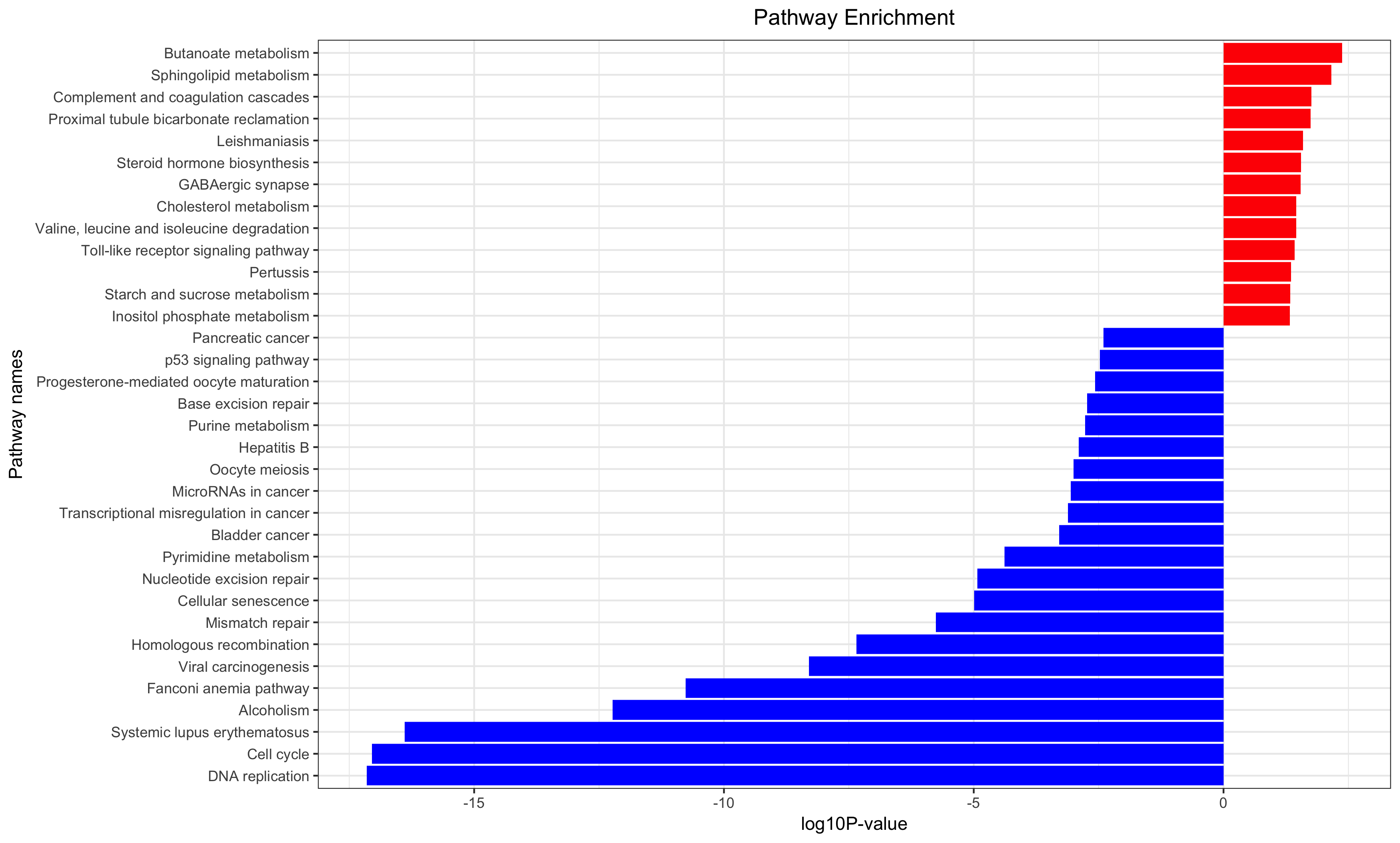
生存分析是补充,只有样本量足够,而且样本的临床信息足够才能做这样的分析。
KM and cox ,需要掌握统计学原理。
算法是亮点,代码见文件夹
算法是门槛,代码见文件夹
不一一介绍,请至少花费几十个小时学习这些代码:
All the videos are uploaded in YouTube: https://www.youtube.com/channel/UC67sImqK7V8tSWHMG8azIVA/videos
如果你在中国,你可能会喜欢B站: https://www.bilibili.com/read/cv719181 ,视频链接: https://www.bilibili.com/video/av26731585/
其实不止是针对转录组表达芯片的数据分析教材,还有转录组数据处理流程,希望你可以仔细看,还有批量生存分析等各种其它统计分析方法我也会慢慢添加。
主要是根据大家的需求啦,希望大家多多反馈和提问哈!
如果你觉得我的教程对你有帮助,请赞赏一杯咖啡哦!
如果你的赞赏超过了50元,请在扫描赞赏的同时留下你的邮箱地址,我会发送给你一个惊喜哦!

关于我们
- 我的博客:生信菜鸟团 http://www.bio-info-trainee.com/
- 我们的论坛:生信技能树 http://www.biotrainee.com/thread-1376-1-1.html
- 我们的VIP社区:https://vip.biotrainee.com/d/311-
- 我们的微信公众号: https://mp.weixin.qq.com/s/egAnRfr3etccU_RsN-zIlg
- 我们的知识星球: https://t.zsxq.com/VjmQZNn
- 我们的腾讯课堂: https://biotree.ke.qq.com/
- 请善用搜索功能:http://weixin.sogou.com/
- 发邮件向我反馈 jmzeng1314@163.com