You signed in with another tab or window. Reload to refresh your session.You signed out in another tab or window. Reload to refresh your session.You switched accounts on another tab or window. Reload to refresh your session.Dismiss alert
Truvari uses hard thresholds to determine if calls are matching. This makes for simpler code, usability, and understanding of Truvari's decision making. However, there are cases where a hard threshold may not be optimal. For example, if we were to set the sequence similarity threshold to 0.95 via --pctsim 0.95, two 50bp insertion calls at the same position but with 3bp different would have a sequence similarity of 0.94 and would not be considered matching. However, two 100bp insertion calls with upto 5bp differene would be considered matching with this hard threshold.
There are many options for how to handle thresholding. Here, we'll show how to leverage Truvari's 'Multi' annotation as well as explore using Truvari library to 'reclassify' matches using KMeans clustering.
Data Setup
First, we'll use SVs found in ticket #91 and run Truvari using v3.1-dev.
We can plot this data for quick view (See #93 ) and see the orange false-negative (fn) as well as some red false-positive (fp) matches that are grouped near the mass of green true-positives (tp). A number of these false calls could easily become true with lower thresholds. By using --multimatch we could also raise the number of TPs. We can check this by using the Multi tag added to false calls. Multi is added to false positives/negatives that pass the thresholds, but were 'beaten' by a better match. Using the vcf2df DataFrame:
--multimatch would have matched 6 more false negatives and 138 more false positives. Allowing flexibility beyond 1-to-1 matching increases performance, but we're still potentially missing matches that are only just below one of the thresholds.
KMeans clustering
To make a KMeans clusterer, we'll leverage the Truvari library to pull out all the matching metrics for every call pair.
importpysamimportnumpyasnpimportpandasaspdimportseabornassbimporttruvariimportmatplotlib.pyplotaspltfromsklearnimportcluster# Setup the input filesref=pysam.FastaFile("human_g1k_v37.fa")
base=pysam.VariantFile("HG002_SVs_Tier1_v0.6.vcf.gz")
comp=pysam.VariantFile("cutesv_filtered_sorted.vcf.gz")
reg=truvari.RegionVCFIterator(base, comp, "HG002_SVs_Tier1_v0.6.bed", 50000)
# Build a matcher and set the parameters. We'll just use defaultsmatcher=truvari.Matcher()
matcher.params.reference=ref# Use Truvari to get all the matching metricschunks=truvari.bench.chunker(matcher, ('base', reg.iterate(base)), ('comp', reg.iterate(comp)))
matches= []
forresultinmap(truvari.bench.compare_chunk, chunks):
formatchinresult:
# Other metrics are available (e.g. start distance `st_dist`)# But we'll just cluster on thesematches.append([match.ovlpct, match.seqsim, match.sizesim])
cols= ["ovlpct", "seqsim", "sizesim"]
matches=pd.DataFrame(matches, columns=cols)
# Many calls have no neighboring calls, and therefore Nones. We'll fill those in as zeros.matches.fillna(0, inplace=True)
# perform the clusteringclust=cluster.KMeans(2, random_state=2)
clust.fit(matches)
labels=pd.Series(clust.labels_, name="label", index=matches.index, dtype=str)
matches["label"] =labels# Look at the cluster centersprint(clust.cluster_centers_)
Reclassifying Data
Here we're clustering on all possible pairs found given the --chunksize, and because we didn't try to preserve any of the identifiers of variants during our map, we'll now need to reuse clust to reclassify our original results/data.jl from the 'hard threshold' to our KMeans cluster
# load the original resultdata=joblib.load("results/data.jl")
to_predict=data[["PctRecOverlap", "PctSeqSimilarity", "PctSizeSimilarity"]].fillna(0)
# predict their clusternew_labels=clust.predict(to_predict.values.astype(float))
data["new_state_label"] =pd.Series(new_labels, index=data.index)
Summarizing Results
Let's inspect the difference our new dynamic thresholding makes for our results.
# make a summarysummary=data.groupby(["state", "new_state_label"]).size().unstack()
Our summary shows that we're relabeling a number of false calls from 0 to 1, where here 0 is our KMeans cluster 'false' an 1 is True. (Note: Visual inspection tells us that the KMeans assigns 1 to matching pairs and 0 to non-matching. This may not always be the case.) Our false negatives are lower by ~1/2 and the false positives by ~1/3. What's interesting is how we're getting these changes without losing any true-positive calls despite not building the KMeans cluster with any information about what separates a True from a False call.
We can also recalculate the precison/recall numbers with some simple data manipulation.
While this may only be a marginal increase (F1 higher by 1.5p.p.), it does show how softer thresholding can improve performance.
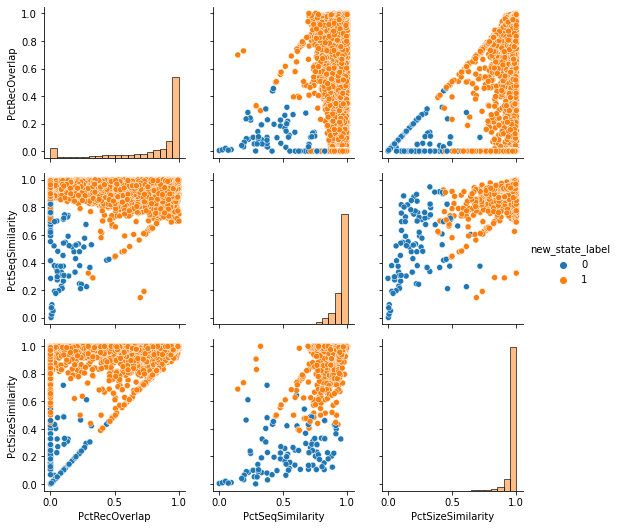
And finally, we'll look at PairPlot of the features with the new labeling. In this updated figure, we can see a better separation between our true and false matches.
Caveats
Again, we may have been able to get these performance increases by simply lowering some of the thresholds or by running Truvari initially by using --multimatch. Reclassifying in this way is like using multimatch because the KMeans prediction has no knowledge of if a call is used more than once.
It's also important to note that KMeans clustering may not be the best approach here and you should explore other ways to make this dynamic threshold. In fact, I would caution against blindly using this approach because SV results can have such different resolutions. Without care, one could easily over or under cluster their results and misrepresent a call set's performance; not to mention how this result has a huge class-imbalance and likely other considerations. But this does illustrate how Truvari can help facilitate more sophisticated matching.
While there may not be a single "best" way to do thresholding, we'll keep working to find a balanced approach and figure out how to integrate it into the tool while preserving Truvari's core usability, code readability, and library reusability.
reacted with thumbs up emoji reacted with thumbs down emoji reacted with laugh emoji reacted with hooray emoji reacted with confused emoji reacted with heart emoji reacted with rocket emoji reacted with eyes emoji
-
Dynamic Matching
Truvari uses hard thresholds to determine if calls are matching. This makes for simpler code, usability, and understanding of Truvari's decision making. However, there are cases where a hard threshold may not be optimal. For example, if we were to set the sequence similarity threshold to 0.95 via
--pctsim 0.95, two 50bp insertion calls at the same position but with 3bp different would have a sequence similarity of 0.94 and would not be considered matching. However, two 100bp insertion calls with upto 5bp differene would be considered matching with this hard threshold.There are many options for how to handle thresholding. Here, we'll show how to leverage Truvari's 'Multi' annotation as well as explore using Truvari library to 'reclassify' matches using KMeans clustering.
Data Setup
First, we'll use SVs found in ticket #91 and run Truvari using v3.1-dev.
truvari bench --passonly -b HG002_SVs_Tier1_v0.6.vcf.gz \ --includebed HG002_SVs_Tier1_v0.6.bed \ -c cutesv_filtered_sorted.vcf.gz \ -o results \ -f human_g1k_v37.fa truvari vcf2df -b -i results/ results/data.jlWe can plot this data for quick view (See #93 ) and see the orange false-negative (fn) as well as some red false-positive (fp) matches that are grouped near the mass of green true-positives (tp). A number of these false calls could easily become true with lower thresholds. By using
--multimatchwe could also raise the number of TPs. We can check this by using theMultitag added to false calls.Multiis added to false positives/negatives that pass the thresholds, but were 'beaten' by a better match. Using thevcf2dfDataFrame:--multimatchwould have matched 6 more false negatives and 138 more false positives. Allowing flexibility beyond 1-to-1 matching increases performance, but we're still potentially missing matches that are only just below one of the thresholds.KMeans clustering
To make a KMeans clusterer, we'll leverage the Truvari library to pull out all the matching metrics for every call pair.
Reclassifying Data
Here we're clustering on all possible pairs found given the
--chunksize, and because we didn't try to preserve any of the identifiers of variants during ourmap, we'll now need to reuseclustto reclassify our originalresults/data.jlfrom the 'hard threshold' to our KMeans clusterSummarizing Results
Let's inspect the difference our new dynamic thresholding makes for our results.
Our summary shows that we're relabeling a number of false calls from 0 to 1, where here 0 is our KMeans cluster 'false' an 1 is True. (Note: Visual inspection tells us that the KMeans assigns 1 to matching pairs and 0 to non-matching. This may not always be the case.) Our false negatives are lower by ~1/2 and the false positives by ~1/3. What's interesting is how we're getting these changes without losing any true-positive calls despite not building the KMeans cluster with any information about what separates a True from a False call.
We can also recalculate the precison/recall numbers with some simple data manipulation.
While this may only be a marginal increase (F1 higher by 1.5p.p.), it does show how softer thresholding can improve performance.
And finally, we'll look at PairPlot of the features with the new labeling. In this updated figure, we can see a better separation between our true and false matches.
Caveats
Again, we may have been able to get these performance increases by simply lowering some of the thresholds or by running Truvari initially by using
--multimatch. Reclassifying in this way is like using multimatch because the KMeans prediction has no knowledge of if a call is used more than once.It's also important to note that KMeans clustering may not be the best approach here and you should explore other ways to make this dynamic threshold. In fact, I would caution against blindly using this approach because SV results can have such different resolutions. Without care, one could easily over or under cluster their results and misrepresent a call set's performance; not to mention how this result has a huge class-imbalance and likely other considerations. But this does illustrate how Truvari can help facilitate more sophisticated matching.
While there may not be a single "best" way to do thresholding, we'll keep working to find a balanced approach and figure out how to integrate it into the tool while preserving Truvari's core usability, code readability, and library reusability.
Beta Was this translation helpful? Give feedback.
All reactions