Home
Molecular heterogeneities bring great challenges for cancer diagnosis and treatment. Recent advance in single cell RNA-sequencing (scRNA-seq) technology make it possible to study cancer transcriptomic heterogeneities at single cell level.
Here, we develop an R package named scCancer which focuses on processing and
analyzing scRNA-seq data for cancer research. Except basic data processing steps,
this package takes several special considerations for cancer-specific features.
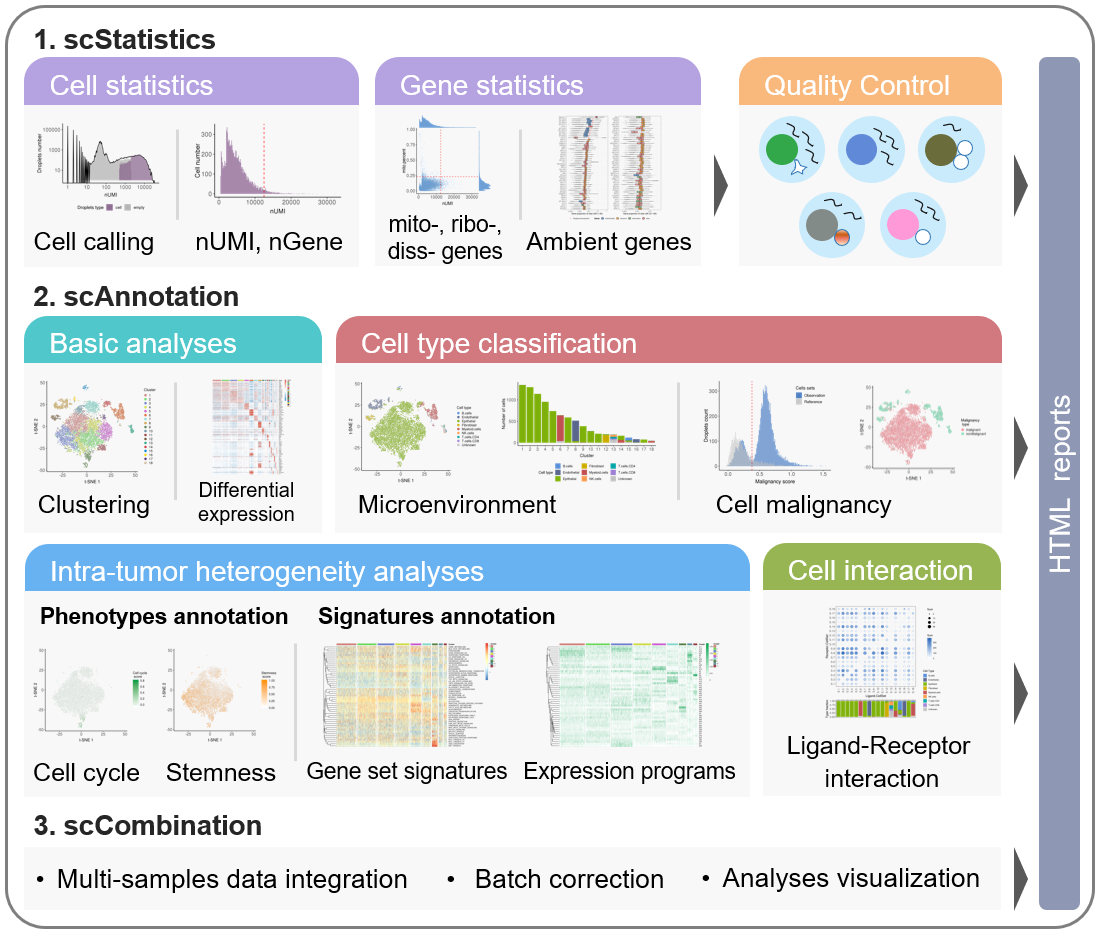
The workflow of scCancer mainly consists of two parts: scStatistics and scAnnotation.
- The
scStatisticsperforms basic statistical analysis of raw data and quality control. - The
scAnnotationperforms functional data analyses and visualizations, such as low dimensional representation, clustering, cell type classification, malignancy estimation, cellular phenotype scoring, gene signature analysis, etc.
After these analyses, user-friendly graphic reports will be generated.
- Memery: >= 32G (for a data with ~10000 cells)
- R version: >= 3.5.0
Firstly, please install or update the package devtools by running
install.packages("devtools")Then the scCancer can be installed via
library(devtools)
devtools::install_github("wguo-research/scCancer")library(scCancer)The scCancer is mainly designed for 10X Genomics platform,
and it requires a data folder containing the results generated by the software
Cell Ranger.
In general, the data folder needs to be organized as following which is the output of Cell Ranger V3:
/sampleFolder
├── filtered_feature_bc_matrix
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── raw_feature_bc_matrix
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
└── web_summary.html
Comparing to Cell Ranger V2 (CR2), Cell Ranger V3 (CR3) can identify cells with
low RNA content better. So we suggest to use CR3 to do alignment and cell-calling.
Considering that some open data is from CR2 or the raw matrix isn't supported, we
specially deisgn the pipeline to be compatible with these situations.
A common structure of CR2 is as below.
/sampleFolder
├── filtered_gene_bc_matrices
│ └── hg19
│ ├── barcodes.tsv
│ ├── genes.tsv
│ └── matrix.mtx
├── raw_gene_bc_matrices
│ └── hg19
│ ├── barcodes.tsv
│ ├── genes.tsv
│ └── matrix.mtx
└── web_summary.html
For other droplet-based platforms, the data folder can be prepared likewise.
Here, we provide an example data of
kidney cancer from 10X Genomics. Users can download it and run following scripts
to understand the workflow of scCancer. And following are the links of generated HTML reports:
The scStatistics mainly implements quality control for the expression matrix
and return some suggested thresholds to filter cells and genes.
Following is the example script to run the first step scStatistics.
And using help(runScStatistics) can get more details about the arguments
of runScStatistics to realize personalized setting.
library(scCancer)
# A path containing the cell ranger processed data
dataPath <- "./data/KC-example"
# A path used to save the results files
savePath <- "./results/KC-example"
# The sample name
sampleName <- "KC-example"
# The author name or a string used to mark the report.
authorName <- "G-Lab@THU"
# Run scStatistics
stat.results <- runScStatistics(
dataPath = dataPath,
savePath = savePath,
sampleName = sampleName,
authorName = authorName
)Running the scStatistics script will generate some files as below:
- report-scStat.html : A HTML report containing all results and corresponding description.
- report-scStat.md : A markdown report.
- figures/ : All figures generated during the pipeline.
- report-figures/: All figures presented in the report.
- cellManifest-all.txt : The results calculated for all droplets.
- cell.QC.thres.txt : The suggested thresholds to filter poor-quality cells.
- geneManifest.txt : The results calculated for each genes.
-
report-cellRanger.html: The summary report generated by
Cell Ranger.
Using the QC thresholds, the scAnnotation filters cells and genes firstly, and then
performs basic operations (normalization, log-transformation, highly variable genes identification,
removing unwanted variance, scaling, centering, dimension reduction, clustering,
and differential expression analy-sis) using R package Seurat V3[1,2].
Besides, scAnnotation also performs some cancer-specific analyses:
-
Cancer micro-environmental cell type classification : In this step, we develop a data-driven model (one-class logistic regression) to predict cell types, including endothelial cells, fibroblast, and immune cells (CD4+ T cells, CD8+ T cells, B cells, nature killer cells, and myeloid cells).
-
Cell malignancy estimation : In this step, we refer to the algorithm of R package
infercnvto estimate an initial CNV profiles [3]. Then, we take advantage of cells’ neighborhoods information to smooth CNV values and define the malignancy score as the mean of the squares of them. -
Cell cycle analysis : In this step, to analyze intra-tumor cell phenotype heterogeneity, we define cell cycle score with the relative average expression of a list of G2/M and S phase markers, by using the function “AddModuleScore” of Seurat [2].
-
Cell stemness analysis : In this step, to analyze intra-tumor cell phenotype heterogeneity, we define cell stemness score with the Spearman correlation coefficient between cells’ expression and our pre-trained stemness signature, by referring to the algorithm of Malta et al [4].
-
Gene set signature analysis : In this step, we provide two methods to calculated gene set signature scores: GSVA and relative average expression levels. By default, we use 50 hallmark gene sets from MSigDB.
-
Expression programs identification : In this step, we use non-negative matrix factorization (NMF) to unsupervised identify potential expression program signatures.
Following is the example script to run the second step scAnnotation.
And using help(runScAnnotation) can get more details about the arguments
of runScAnnotation to realize personalized setting.
library(scCancer)
# A path containing the cell ranger processed data
dataPath <- "./data/KC-example"
# A path containing the scStatistics results
statPath <- "./results/KC-example"
# A path used to save the results files
savePath <- "./results/KC-example"
# The sample name
sampleName <- "KC-example"
# The author name or a string used to mark the report.
authorName <- "G-Lab@THU"
# Run scAnnotation
anno.results <- runScAnnotation(
dataPath = dataPath,
statPath = statPath,
savePath = savePath,
authorName = authorName,
sampleName = sampleName,
geneSet.method = "average" # or "GSVA"
)Running the scAnnotation script will generate some files/folders as below:
- report-scAnno.html : A HTML report containing all results and corresponding description.
- report-scAnno.md : A markdown report.
- figures/ : All figures generated during the pipeline.
- report-figures/: All figures used in the report.
- gene.manifest.filter.txt : The filtered genes during QC.
- expr.RDS : A Seurat object.
- diff.expr.genes/ : Differentially expressed genes information for all clusters.
- cellAnnotation.txt : The annotation results for each cells.
- malignancy/: All results of cell malignancy estimation.
- expr.programs/: All results of expression programs identification.
After the step scStatistics, a HTML report (report-scStat.html) will be generated, which persents the statistical features of the data from various perspectives (nUMI, nGene, mito.percent, ribo.percent, diss.percent).
By identifying outliers from the distribution of these metrics, scCancer adaptively provides some suggested thresholds to filter low-quality cells and records them in file cell.QC.thres.txt.
Besides, users can also modify the values in file cell.QC.thres.txt and then the step scAnnotation will use the updated thresholds to perform cell QC and downstream analyses.
For the step gene QC, the scCancer filters genes according to three aspects of information.
- First, it filters mitochondrial, ribosomal, and dissociation-associated genes.
- Second, it filters genes with the number of expressed cells less than argument
nCell.min(the default is 3). - Third, it filters genes with the background percentage larger than the argument
bgPercent.max(the default is 0.001). The distribution ofbgPercent.maxcan be found in thereport-scStat.html.
Users can also modify the value of these arguments according to their needs.
For patient-drived tumor xenograft (PDX) samples, it contains both human and mouse cells generally. In order to deal with these human-mouse-mixed data, users can explicitly set the arguments hg.mm.mix, species, hg.mm.thres, and mix.anno of relevant functions.
More details for the meaning of these arguments can be found by using command help().
If the sample data is generated by Cell Ranger V2 (CR2) and contain 'raw_gene_bc_matrices' matrix, the scCancer package can re-perform cell calling using
the method EmptyDrop (the name of R package is DropletUtils). But users need to manually install the DropletUtils and import it in script.
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("DropletUtils")
library(DropletUtils)[1] Butler A, Hoffman P, Smibert P, et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species[J]. Nature biotechnology, 2018, 36(5): 411.
[2] Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data[J]. Cell, 2019.
[3] inferCNV of the Trinity CTAT Project. https://github.com/broadinstitute/inferCNV
[4] Malta T M, Sokolov A, et al. Machine learning identifies stemness features asso-ciated with oncogenic dedifferentiation. Cell, 2018, 173(2): 338-354. e15.
Here is the output of sessionInfo() on the system.
R version 3.6.0 (2019-04-26)
Platform: x86_64-conda_cos6-linux-gnu (64-bit)
Running under: Ubuntu 16.04.2 LTS
Matrix products: default
locale:
[1] en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] scCancer_1.0.0
loaded via a namespace (and not attached):
[1] Seurat_3.1.0 Rtsne_0.15 colorspace_1.4-1
[4] ggridges_0.5.1 IRdisplay_0.7.0 markdown_1.1
[7] base64enc_0.1-3 leiden_0.3.1 listenv_0.7.0
[10] npsurv_0.4-0 bit64_0.9-7 ggrepel_0.8.1
[13] AnnotationDbi_1.46.1 codetools_0.2-16 splines_3.6.0
[16] R.methodsS3_1.7.1 lsei_1.2-0 geneplotter_1.62.0
[19] knitr_1.24 shinythemes_1.1.2 zeallot_0.1.0
[22] IRkernel_1.0.1.9000 jsonlite_1.6 annotate_1.62.0
[25] ica_1.0-2 cluster_2.0.9 png_0.1-7
[28] R.oo_1.22.0 pheatmap_1.0.12 graph_1.62.0
[31] uwot_0.1.3 shiny_1.3.2 sctransform_0.2.0
[34] compiler_3.6.0 httr_1.4.1 backports_1.1.4
[37] assertthat_0.2.1 Matrix_1.2-17 lazyeval_0.2.2
[40] later_0.8.0 htmltools_0.3.6 tools_3.6.0
[43] rsvd_1.0.2 igraph_1.2.4.1 gtable_0.3.0
[46] glue_1.3.1 RANN_2.6.1 reshape2_1.4.3
[49] dplyr_0.8.3 Rcpp_1.0.2 Biobase_2.44.0
[52] vctrs_0.2.0 gdata_2.18.0 ape_5.3
[55] nlme_3.1-140 gbRd_0.4-11 lmtest_0.9-37
[58] xfun_0.9 stringr_1.4.0 globals_0.12.4
[61] miniUI_0.1.1.1 mime_0.7 lifecycle_0.1.0
[64] irlba_2.3.3 gtools_3.8.1 XML_3.98-1.20
[67] NNLM_0.4.3 future_1.14.0 MASS_7.3-51.4
[70] zoo_1.8-6 scales_1.0.0 promises_1.0.1
[73] parallel_3.6.0 RColorBrewer_1.1-2 memoise_1.1.0
[76] reticulate_1.13 pbapply_1.4-2 gridExtra_2.3
[79] ggplot2_3.2.1 ggExtra_0.9 RSQLite_2.1.2
[82] stringi_1.4.3 GSVA_1.32.0 highr_0.8
[85] S4Vectors_0.22.0 caTools_1.17.1.2 BiocGenerics_0.30.0
[88] bibtex_0.4.2 repr_1.0.1 Rdpack_0.11-0
[91] SDMTools_1.1-221.1 rlang_0.4.0 pkgconfig_2.0.2
[94] bitops_1.0-6 evaluate_0.14 lattice_0.20-38
[97] ROCR_1.0-7 purrr_0.3.2 labeling_0.3
[100] htmlwidgets_1.3 bit_1.1-14 cowplot_1.0.0
[103] tidyselect_0.2.5 GSEABase_1.46.0 RcppAnnoy_0.0.12
[106] plyr_1.8.4 magrittr_1.5 R6_2.4.0
[109] IRanges_2.18.1 gplots_3.0.1.1 DBI_1.0.0
[112] pbdZMQ_0.3-3 pillar_1.4.2 fitdistrplus_1.0-14
[115] survival_2.44-1.1 RCurl_1.95-4.12 tibble_2.1.3
[118] future.apply_1.3.0 tsne_0.1-3 crayon_1.3.4
[121] uuid_0.1-2 KernSmooth_2.23-15 plotly_4.9.0
[124] grid_3.6.0 data.table_1.12.2 blob_1.2.0
[127] diptest_0.75-7 metap_1.1 digest_0.6.20
[130] xtable_1.8-4 tidyr_1.0.0 httpuv_1.5.2
[133] R.utils_2.9.0 stats4_3.6.0 RcppParallel_4.4.3
[136] munsell_0.5.0 viridisLite_0.3.0