Prepare 3 tab format for RNAseq dataset uploading
The following R code goes through preparing a dataset for upload to gEAR using the 3 tab format. If you wish for assistance with the formatting or upload process, please contact our curator team (https://umgear.org/contact.html)
- load in count file
countsFileName="count.txt"
count = read.delim(sep="\t",countsFileName, header=T, check.names=F)
names(count)[1] = "gene"
rownames(count) = count$gene
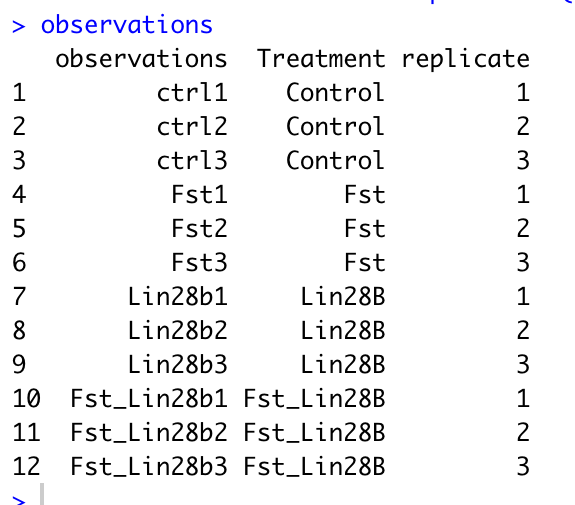
- generate the observation file (this can be done either via code or creating a tab delimited file in Excel.
options(repr.matrix.max.rows=50, repr.matrix.max.cols=200)
meta = read.delim("phenotype.txt")
#re-name the columns, change gender to your variable of interest
names(obs) = c("observations","genotype", "gender")Below is an example observation file. The first column must be named observations with rows corresponding to your sample names. You can add any additional columns that you would like to be able to use for comparison in gEAR. If you have replicate samples, to enable all the functionalities of the statistical tools in gEAR, please include a column with the name 'replicate' and values similar to below. A column named "replicate", “BioRep”, or “TechRep” is needed for conducting statistic analysis with the comparison tool. The code for adding a replicate column is below, just change the names in the group_by function to match your own column names.
# Adding a replicate column to your dataframe
# Change the variables (e.g. genotype, gender)in group_by to those in your own dataframe.
obs <- obs %>% group_by(genotype, gender) %>% mutate(replicate = row_number())
obs = data.frame(obs)
write.table(obs, file= "observation.tab", sep="\t", row.names=F, quote=F)- annotation. check whether there is ensemble in the count file
temp=count[grepl("ENS",count[,1]),]
if (nrow(temp)>0) {
print('ensembl ID are detected')
} else {
print('Error: no ensemble detected')}Here we are using the mouse annotation database, if you wish to use a different database run listDatasets(mart) to get a full list of available annotations.
library(biomaRT)
mart = useMart( 'ensembl' )
datasets <- listDatasets(mart)
mart = useDataset( 'mmusculus_gene_ensembl' , mart = mart )
ensembl = getBM( attributes = c('ensembl_gene_id','external_gene_name') , mart=mart)
names(ensembl)[2] = "gene_symbol"
head(ensembl)
#only keep the first ensemble ID for each gene
ensembl.m=ensembl[!duplicated(ensembl$gene_symbol),]
#merge by gene_symbol or ensembl ID
count.ann = merge(ensembl, count, by.x="gene_symbol", by.y="gene_symbol")
names(count.ann)[c(2,1)] = c("ensembl_ID", "gene_symbol")
write.table(count.ann[-1], file= "expression.tab", sep="\t", quote=F, row.names=F)
genes_to_use=count.ann[c(2,1)]
write.table(genes_to_use,file= "genes.tab", sep="\t", quote=F,row.names=F)if the count file is raw count, basic normalization need to be completed before uploading
count_to_norm=count.ann[-c(1:2)]
row.names(count_to_norm)=count.ann[,1]
cpm=sweep(count_to_norm,2,colSums(count_to_norm),FUN="/")*10^6
##quantile normalization of cpm
quantile_normalisation <- function(df){
df_rank <- apply(df,2,rank,ties.method="min")
df_sorted <- data.frame(apply(df, 2, sort))
df_mean <- apply(df_sorted, 1, mean)
index_to_mean <- function(my_index, my_mean){
return(my_mean[my_index])}
df_final <- apply(df_rank, 2, index_to_mean, my_mean=df_mean)
rownames(df_final) <- rownames(df)
return(df_final)}
nq=quantile_normalisation(data.frame(cpm))
write.table(nq, file=paste(outfile_prefix, "expression.tab", sep = ""), sep="\t", quote=F, row.names=T)Compress files together for uploading
system( 'tar -czvf upload.tar.gz *.tab')