Allow 2 Variant_Classification shown on Oncoplot #1062
Comments
|
Hi, Thanks for the issue. Unfortunately, it is not possible to do so. Also, if there are more than two events (e.g, two mutations and a CN change i.e, complex event) - it becomes even harder. |
|
hi, I just add
The oncoplot vignette is here: https://yunuuuu.github.io/ggalign/dev/articles/oncoplot.html |
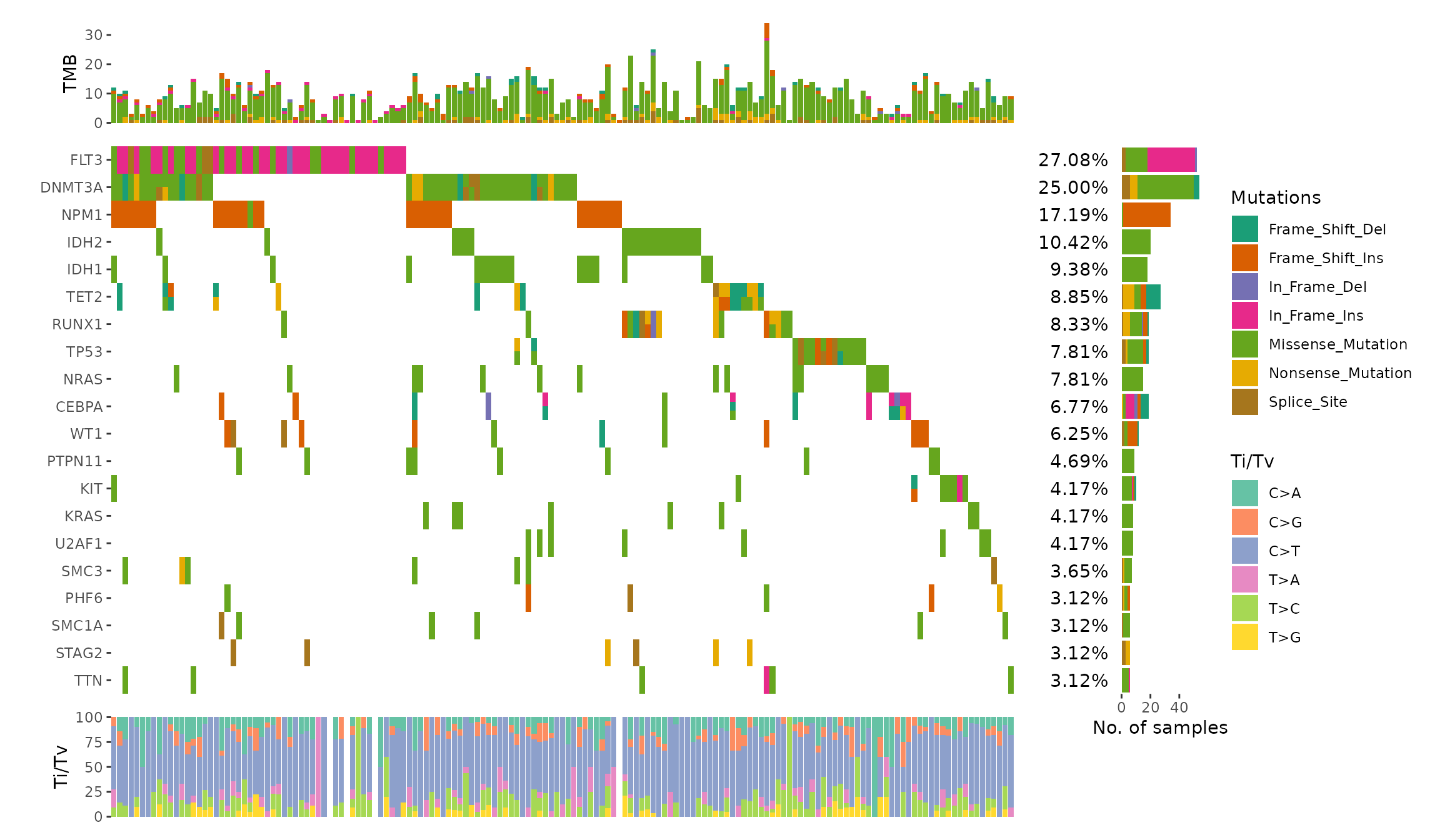
|
Hi @Yunuuuu That's pretty cool!! Nicely done. I will mention this in the vignette so that many people can find it. |
|
***@***.*** appreciated! I just wonder with ggalign to plot MAF obj, will CNV either within OR separate from main plot could be also compatible with maftools? To show more than 1 variant per sample per gene is quite useful as well as CNV data as well. Not sure fully compatible? Thanks for additional feature!Sent from my iPhoneOn Nov 16, 2024, at 11:50, Anand Mayakonda ***@***.***> wrote:
Hi @Yunuuuu
That's pretty cool!! Nicely done. I will mention this in the vignette so that many people can find it.
—Reply to this email directly, view it on GitHub, or unsubscribe.You are receiving this because you authored the thread.Message ID: ***@***.***>
|
|
Unfortunately, it is not. |
|
Thanks for the info. And ggoncoplot() seems don’t have flexibility to sort genes manually or samples according to annotation or specific sample order? How to achieve this with ggoncoplot(). I know maftools can be done with provided arguments.
…Sent from my iPhone
On Nov 16, 2024, at 12:08, Anand Mayakonda ***@***.***> wrote:
Unfortunately, it is not.
The more variant types - the plot becomes more complicated. Hence oncoplot shows them as Multi_Hit instead (two or more mutations). There are some cases with >2 mutations and copy number changes - those will simply be annotated as complex_event.
—
Reply to this email directly, view it on GitHub<#1062 (comment)>, or unsubscribe<https://github.com/notifications/unsubscribe-auth/BHAFOAPZRADLRRPGJJBJ6LD2A53XDAVCNFSM6AAAAABQZX3Z7WVHI2DSMVQWIX3LMV43OSLTON2WKQ3PNVWWK3TUHMZDIOBQGY3DQNRYGA>.
You are receiving this because you authored the thread.Message ID: ***@***.***>
The information transmitted in this electronic communication is intended only for the person or entity to whom it is addressed. It may also contain confidential and/or privileged material. Any use of the information transmitted by persons or entities other than the intended recipient is strictly prohibited. If this email was sent to you in error and includes sensitive Dana-Farber information (for example, patient information, Social Security Numbers, credit card information), please communicate this to Dana-Farber's Compliance Hotline at www.compliancereport.org <http://www.compliancereport.org> . If the information transmitted does not contain such sensitive information, please contact the sender and properly dispose of the email.
Please note that this e-mail is not secure (encrypted). If you do not wish to continue communication over unencrypted e-mail, please notify the sender of this message immediately. Continuing to send or respond to e-mail after receiving this message means you understand and accept this risk and wish to continue to communicate over unencrypted e-mail.
|
Now, ggalign only implement methods for
Maybe you need |
|
Cool, I will take a look. And as for annotations for the samples, I will use ggalign_attr() with argument “sample_anno” that are within MAF obj? How to choose specific annotation columns of from all annotations available one by one so u can customize it’s color etc? (E.g. I just want gender, MRD_status, IGHV_status one by one, and those I can check their existence by getClinicalData() function from maftools). Thanks for suggestions.
Sent from my iPhone
On Nov 16, 2024, at 12:16, Yunuuuu ***@***.***> wrote:
External Email - Use Caution
@wangzunq<https://secure-web.cisco.com/1WyssOpXUJFofoR54rpyZFvZc38tizF88ZegTdKxf6EdO8HkGc6kbOgGDbiVt4cQiqfE-XsChGfx69IvFG3wRMDMrMyOsEsiNEHOXuW4KZ-SJ3v2LTOkA-N0fJoWHx8pl5fLSlSWBni43FTowmsvFYJIXt_zcKTpu7RO6Y4jx-XwUB5sdFNv4xl5TIylB5vYJEC2jT_Pv8wBwXouuzWPYbzVoOTGzC-ARSBftveAS-29joyec9HI0vEdo5R2-THNfSgcpwa1CvCtTUm9A-hMaSaksQ0sqGFEQX5mznkbXMUwjD5DHgf5IcMNxj2-TC_qX7A34usV6z6vLFM4fi1rEGQ/https%3A%2F%2Fgithub.com%2Fwangzunq>
I just wonder with ggalign to plot MAF obj, will CNV either within OR separate from main plot could be also compatible with maftools? To show more than 1 variant per sample per gene is quite useful as well as CNV data as well. Not sure fully compatible?
Now, ggalign only implement methods for MAF object, but it easy to create methods for all objects in the maftoolds package.
ggoncoplot() seems don’t have flexibility to sort genes manually or samples according to annotation or specific sample order?
Maybe you need align_order(), please see link: https://yunuuuu.github.io/ggalign/dev/articles/layout-customize.html#align_order<https://secure-web.cisco.com/1u43K5E0IaHmJ8VR1Y4rxOVndEzXxjucnMVc0QcEJvEbH_BVYuQGvZfxll1mdLC0Qi47uZoE2ThfFuANIccQla-RjlN_hCOBOvk9awh6FTuVhROiuWjVPmHxjzVOk1wD-ulMRHDrqdIiRGUxj6DtYdLNxdjqMcluBuWaZ29-WuJASuKMkydIuOJfccEmGmLLrvrT2nynDEESU4RcM1MsybUG6zFyih0UcwIx28FwPQsBcW1sBOkhqrq3iZ-scebJOjmyhkMD3hg5UCXT2ETR59DxVjA33KdvrRS8N_9a4QGFf-LPiAGCmylo5xAJsGhuaI5XTW8a-58sACreAQyiuXQ/https%3A%2F%2Fyunuuuu.github.io%2Fggalign%2Fdev%2Farticles%2Flayout-customize.html%23align_order>. it allows the usa of ordering character or integer index.
—
Reply to this email directly, view it on GitHub<https://secure-web.cisco.com/1ja8ybW8HSULRMWVrEiQhN_UQaX8iP22HESXhB86jBG40rNmfcS-oGBofN8pNJgxQQ7EX3zeyjqwfKrphPf7y6WyRrnFWQl8JhJZG49LbEEOqU-cwVKqMUbQZZXAFgi1uv-ciAhrjsWepWNHyt4RkKCOjcrEn0Xdwlx0cpeOFSTmx6-9PwtWRcZ1RqnR0a5Z3p1rKdMbIAMLX8YxQEbC1rI047MQ0Y-Dmu0Fm92hu0fBmFfPbDm4SUFvzfIdJlGWxWSNzah2th3VZ29pJz_-WBHI5DG3JszSN_Bd8AnnOBSeTPwuAxxqnvaTjHiVr4jKhLt6R2jR904iVNq4cXP7Bqw/https%3A%2F%2Fgithub.com%2FPoisonAlien%2Fmaftools%2Fissues%2F1062%23issuecomment-2480671831>, or unsubscribe<https://secure-web.cisco.com/135Cq15CR4kkTFPoWygqD8TJ0iQ-M8cwtSatVqw6JVQ9a_9JEb8Pl8wOwtvJcpL86zC65MCRak1i-IuHOk7FlpQmkaRjeB0SfiUFdcBhA9yN0cGnhGyYPacffykmhCW2owzAyZqOwJy6Sa6O0_yuVjgqGVWYIMJTBrp8MaEQXj_qnzpWoCAMdAEqng8O7Y3ZBygBHUuD4xhTlt016MgHt4dJiGAYEt-_6QoHMcxZXvOre26GOgj4FKmtTDtPPzj_AG9wYrv3k96DIL-uJ5JVrvEFNOihOsxCyiu2twqedTE2gXHRMZy96IdPrt8gOXGbVhggPZB2ji0qzC0XvSx4aLQ/https%3A%2F%2Fgithub.com%2Fnotifications%2Funsubscribe-auth%2FBHAFOALHQ2C5WEGNX2XZ3I32A54WTAVCNFSM6AAAAABQZX3Z7WVHI2DSMVQWIX3LMV43OSLTON2WKQ3PNVWWK3TUHMZDIOBQGY3TCOBTGE>.
You are receiving this because you were mentioned.Message ID: ***@***.***>
The information transmitted in this electronic communication is intended only for the person or entity to whom it is addressed. It may also contain confidential and/or privileged material. Any use of the information transmitted by persons or entities other than the intended recipient is strictly prohibited. If this email was sent to you in error and includes sensitive Dana-Farber information (for example, patient information, Social Security Numbers, credit card information), please communicate this to Dana-Farber's Compliance Hotline at www.compliancereport.org <http://www.compliancereport.org> . If the information transmitted does not contain such sensitive information, please contact the sender and properly dispose of the email.
Please note that this e-mail is not secure (encrypted). If you do not wish to continue communication over unencrypted e-mail, please notify the sender of this message immediately. Continuing to send or respond to e-mail after receiving this message means you understand and accept this risk and wish to continue to communicate over unencrypted e-mail.
|
@PoisonAlien Thank you so much for the kind words! 😊 I'm glad you found it helpful. I'll add full support for all objects in
Absolutely! When there are many samples, distinguishing the smaller tiles can be challenging. That’s why
For more flexibility, users can also opt for In most cases, especially when visualizing the top genes (usually 20–30), Note: |
@wangzunq, Yes, you can use If you want to check the available columns first, you can use Since the lapply(c("gender", "MRD_status", "IGHV_status"), function(x) {
list(
# I presume all these three columns are in the `MAF` object.
ggalign(data = function(data) ggalign_attr(data, "sample_anno")),
# maybe you want to a `geom_tile()` ?
geom_tile(aes(.x, 1L, fill = .data[[x]]))
)
}) |
|
Thank you! This package in conjunction with maftools is so powerful and also appreciate constant improvement on the package. Have a great weekend!
…Sent from my iPhone
On Nov 16, 2024, at 12:42, Yunuuuu ***@***.***> wrote:
External Email - Use Caution
Cool, I will take a look. And as for annotations for the samples, I will use ggalign_attr() with argument “sample_anno” that are within MAF obj? How to choose specific annotation columns of from all annotations available one by one so u can customize it’s color etc? (E.g. I just want gender, MRD_status, IGHV_status one by one, and those I can check their existence by getClinicalData() function from maftools). Thanks for suggestions.
@wangzunq<https://secure-web.cisco.com/1Pzd3lkaYvS0RBPRUXz-hGqeSFl-nRSwdX6hMGpRkSxRsu8lipW7JDqp9YbYGOc8kR1DF4w7nPN17qH9f_9OKniEPmnSpDA1ceoiHmwh5U5D9myOWJBrSMvI2TDEStr-bdSTWOcgK9-L3IMKaod7tNpQHt9egpXMNoNr5BAKQ0_T12Naftv-MKa44ZoJOaKrWaBoo-YNEdfS1uexBJqKOTznjkmbd6BabddzBMDOict07UpDnjAGYgtjHOwnIsVPUaNomvd8N5xdl2J_ObobGBpwzOb6y1klNTvAqCYDNQC_uBBj9hUJWWt9f649H4-FC_WgaIs8jm6vEMy7IwsbtHg/https%3A%2F%2Fgithub.com%2Fwangzunq>, Yes, you can use ggalign_attr() with the argument "sample_anno" to access the sample annotations within your MAF object. To customize specific annotation columns like "gender", "MRD_status", and "IGHV_status", you can use standard ggplot2 syntax to plot the selected columns.
If you want to check the available columns first, you can use getClinicalData() from maftools to get the full set of clinical annotations. But you can always provide data directly to ggalign().
ggalign package is a pure ggplot2 extension, so you can use the same syntax of ggplot2 to add geoms and you can just keep the whole data frame. If you want to add multiple plot simutaneouly, you can add a list (we use lapply to create a series of plots):
lapply(c("gender", "MRD_status", "IGHV_status"), function(x) {
list(
ggalign(data = function(data) ggalign_attr(data, "sample_anno")),
# maybe you want to a `geom_tile()`
geom_tile(aes(.x, 1L, fill = .data[[x]]))
)
})
—
Reply to this email directly, view it on GitHub<https://secure-web.cisco.com/1xA-8C8W0wmRS4U6bf8LkvUW8cbTShnLFGoEKoBHc601NsFz5oxXgZLzupaPZDsRQVSJ_58ZZvcq0hcDbwiR7z9VlCk20bW1OTEl_U-5uzOiuVWTzwY3rphoFpYS_KfUaZWxjsvoUJorMdc1UThpJnlLKISiKB36xydmGn0GZRUueb67qotfaHTdjwOLULnnN2gpi98HJ5YMR8bMcAKbRatx_wAqY4qyHchCl9bPdpl-Yk9pFSiOTX_Qyxre13EA2xL6EatrtM8AKVFNSX04wDe-nOFvKtNRJBLP7tBFlrPDcJ-0X3gd42DbLQUf_vU0PU7MubIqtSSMILBO4yJoytw/https%3A%2F%2Fgithub.com%2FPoisonAlien%2Fmaftools%2Fissues%2F1062%23issuecomment-2480681081>, or unsubscribe<https://secure-web.cisco.com/1G3gIGZQxDyS785NfiLiG9-x_sgpSlm9E90eeYO9e1VjXPl1yIE3H-CViOgQDyd5R0FJDH3i01kLVTS2mR6F3EXS_ftGBjT0j5yUcvPVcJ5RcDJ-AKvTykszE3rHO-NHFkGDb0EcRCSM7FuPhInyFBEoLvWL57TKetpgcF-CLqFEDVkmm3TCiEOR1kmVUO29bMO4aVCky1uIOzHmZsFVJGHammBiK2_UB91heKEca9Luvql7Pjd6R6ROXgiDsNOXYqNg00XBhyvSxyvQpchj8Kp0tTA53cst3dF940tu066O5a62Rx0oIVymq5X_WIQa0qF7L-n1xk5pPmbBBJsK7SA/https%3A%2F%2Fgithub.com%2Fnotifications%2Funsubscribe-auth%2FBHAFOAO4QA44UXPS42JZG5L2A57XZAVCNFSM6AAAAABQZX3Z7WVHI2DSMVQWIX3LMV43OSLTON2WKQ3PNVWWK3TUHMZDIOBQGY4DCMBYGE>.
You are receiving this because you were mentioned.Message ID: ***@***.***>
The information transmitted in this electronic communication is intended only for the person or entity to whom it is addressed. It may also contain confidential and/or privileged material. Any use of the information transmitted by persons or entities other than the intended recipient is strictly prohibited. If this email was sent to you in error and includes sensitive Dana-Farber information (for example, patient information, Social Security Numbers, credit card information), please communicate this to Dana-Farber's Compliance Hotline at www.compliancereport.org <http://www.compliancereport.org> . If the information transmitted does not contain such sensitive information, please contact the sender and properly dispose of the email.
Please note that this e-mail is not secure (encrypted). If you do not wish to continue communication over unencrypted e-mail, please notify the sender of this message immediately. Continuing to send or respond to e-mail after receiving this message means you understand and accept this risk and wish to continue to communicate over unencrypted e-mail.
|
|
In the example, why you subset explicitly as.matrix(data[2:(ncol(data) - 1L)]) for TMB and as.matrix(data[2:7]) for Ti/Tv? How should I inspect those columns for what I want to subset ?
…Sent from my iPhone
On Nov 16, 2024, at 12:42, Yunuuuu ***@***.***> wrote:
Cool, I will take a look. And as for annotations for the samples, I will use ggalign_attr() with argument “sample_anno” that are within MAF obj? How to choose specific annotation columns of from all annotations available one by one so u can customize it’s color etc? (E.g. I just want gender, MRD_status, IGHV_status one by one, and those I can check their existence by getClinicalData() function from maftools). Thanks for suggestions.
@wangzunq<https://github.com/wangzunq>, Yes, you can use ggalign_attr() with the argument "sample_anno" to access the sample annotations within your MAF object. To customize specific annotation columns like "gender", "MRD_status", and "IGHV_status", you can use standard ggplot2 syntax to plot the selected columns.
If you want to check the available columns first, you can use getClinicalData() from maftools to get the full set of clinical annotations. But you can always provide data directly to ggalign().
ggalign package is a pure ggplot2 extension, so you can use the same syntax of ggplot2 to add geoms and you can just keep the whole data frame. If you want to add multiple plot simutaneouly, you can add a list (we use lapply to create a series of plots):
lapply(c("gender", "MRD_status", "IGHV_status"), function(x) {
list(
ggalign(data = function(data) ggalign_attr(data, "sample_anno")),
# maybe you want to a `geom_tile()`
geom_tile(aes(.x, 1L, fill = .data[[x]]))
)
})
—
Reply to this email directly, view it on GitHub<#1062 (comment)>, or unsubscribe<https://github.com/notifications/unsubscribe-auth/BHAFOAO4QA44UXPS42JZG5L2A57XZAVCNFSM6AAAAABQZX3Z7WVHI2DSMVQWIX3LMV43OSLTON2WKQ3PNVWWK3TUHMZDIOBQGY4DCMBYGE>.
You are receiving this because you were mentioned.Message ID: ***@***.***>
The information transmitted in this electronic communication is intended only for the person or entity to whom it is addressed. It may also contain confidential and/or privileged material. Any use of the information transmitted by persons or entities other than the intended recipient is strictly prohibited. If this email was sent to you in error and includes sensitive Dana-Farber information (for example, patient information, Social Security Numbers, credit card information), please communicate this to Dana-Farber's Compliance Hotline at www.compliancereport.org <http://www.compliancereport.org> . If the information transmitted does not contain such sensitive information, please contact the sender and properly dispose of the email.
Please note that this e-mail is not secure (encrypted). If you do not wish to continue communication over unencrypted e-mail, please notify the sender of this message immediately. Continuing to send or respond to e-mail after receiving this message means you understand and accept this risk and wish to continue to communicate over unencrypted e-mail.
|
|
Thanks for the point, I have updated the documents:
Note: |
|
|
|
@wangzunq Thank you so much for the kind words and support! I'm glad you find the new features helpful. I’m constantly working to improve the package, so your feedback means a lot. If you have any suggestions or ideas for further enhancements, feel free to share! |
|
Hello Yunuuuu,
I have posted 2 questions regarding plots. I am wondering they are achievable.
For samples group by cohort and sort for each by mutation frequency, it could be manually done first and supply the sorted samples to param, just wonder if it can be automatically done in the backend?
For Gistic, I am not sure how to proceed with results from different cohort. Please advise. Happy Thanksgiving week.
Best,
Zunqiu
…Sent from my iPhone
On Nov 18, 2024, at 00:52, Yunuuuu ***@***.***> wrote:
@wangzunq<https://github.com/wangzunq> Thank you so much for the kind words and support! I'm glad you find the new features helpful. I’m constantly working to improve the package, so your feedback means a lot. If you have any suggestions or ideas for further enhancements, feel free to share!
—
Reply to this email directly, view it on GitHub<#1062 (comment)>, or unsubscribe<https://github.com/notifications/unsubscribe-auth/BHAFOAKLFZQF5BBCMRI4MWT2BF6ARAVCNFSM6AAAAABQZX3Z7WVHI2DSMVQWIX3LMV43OSLTON2WKQ3PNVWWK3TUHMZDIOBSGAYTGMJZGQ>.
You are receiving this because you were mentioned.Message ID: ***@***.***>
The information transmitted in this electronic communication is intended only for the person or entity to whom it is addressed. It may also contain confidential and/or privileged material. Any use of the information transmitted by persons or entities other than the intended recipient is strictly prohibited. If this email was sent to you in error and includes sensitive Dana-Farber information (for example, patient information, Social Security Numbers, credit card information), please communicate this to Dana-Farber's Compliance Hotline at www.compliancereport.org <http://www.compliancereport.org> . If the information transmitted does not contain such sensitive information, please contact the sender and properly dispose of the email.
Please note that this e-mail is not secure (encrypted). If you do not wish to continue communication over unencrypted e-mail, please notify the sender of this message immediately. Continuing to send or respond to e-mail after receiving this message means you understand and accept this risk and wish to continue to communicate over unencrypted e-mail.
|
|
@wangzunq , Sorry for the delayed reply, I missed the message.
I will export the ordering function for you to use. You can simply apply
the general approach would be to first merge the datasets into one unified set, ensuring you add a column to indicate the origin of each dataset (e.g., cohort A, cohort B). This way, you can proceed as if it's one single cohort. I'm not certain whether |
|
Added by 9a8240952683f75268613c0f2eda657dd5e13f57, you can use |
Describe the issue
Hi,
I would like to show 2 variant classification as 2 triangles(split tile) for same gene if that gene happens to have 2 types of variant classifications in sample sample. Currently as long as a gene in the same sample has more than 2 it just mark it as Multi_Hit. Thanks for any suggestions.
The text was updated successfully, but these errors were encountered: