![]()
SAMStat is an efficient C program to quickly display statistics of large sequence files from next generation sequencing projects. When applied to SAM/BAM files all statistics are reported for unmapped, poorly and accurately mapped reads separately. This allows for identification of a variety of problems, such as remaining linker and adaptor sequences, causing poor mapping. Apart from this SAMStat can be used to verify individual processing steps in large analysis pipelines.
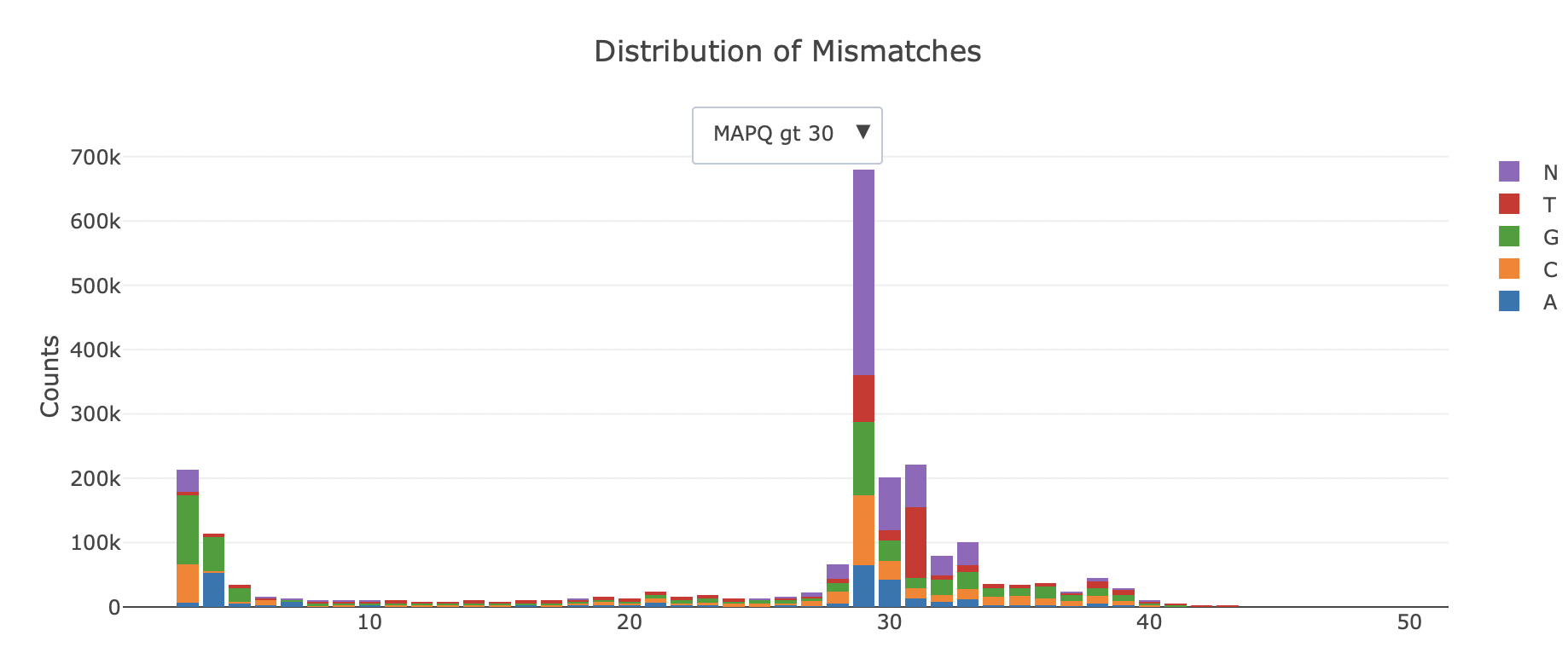
SAMStat reports length distribution, base quality distribution, mapping statistics, mismatch, insertion and deletion error profiles. The output is a single html page:
SAMstat depends on the hdf5 library. To install on linux:
Ubuntu/Debian:
sudo apt-get install -y libhdf5-devOn a mac via brew:
brew install hdf5To build SAMstat:
git clone https://github.com/TimoLassmann/samstat.git
cd samstat
mkdir build
cd build
cmake ..
make
make install samstat <file.sam> <file.bam> <file.fa> <file.fq> ... <options> For each input file SAMStat will create a single html page named after the input file name plus a dot samstat.html suffix.
Available options:
-d/-dir : Output directory. []
NOTE: by default SAMStat will place reports in the same directory as the input files.
-p/-peek : Report stats only on the first <n> sequences. [unlimited]
-t : Number of threads. [4]
will only be used when multiple input files are present.
--plotend : Add base and quality plots relative to the read ends. []
--seed : Random number seed. [0]
--verbose : Enables verbose output. []
-h/-help : Prints help message. []
-v/-version : Prints version information. []Timo Lassmann (2023) “SAMStat 2: quality control for next generation sequencing data.” Bioinformatics. (2023): btad019, https://doi.org/10.1093/bioinformatics/btad019
Lassmann et al. (2010) “SAMStat: monitoring biases in next generation sequencing data.” Bioinformatics 27.1 (2011): 130-131. doi:10.1093/bioinformatics/btq614