IMPORTANT: Do not share the code without my express permission as it is unpublished (manuscript in preparation)
![]()
- Table of content
- Jovian description
- Requirements
- Instructions
- FAQ
- Example Jovian report
- Acknowledgements
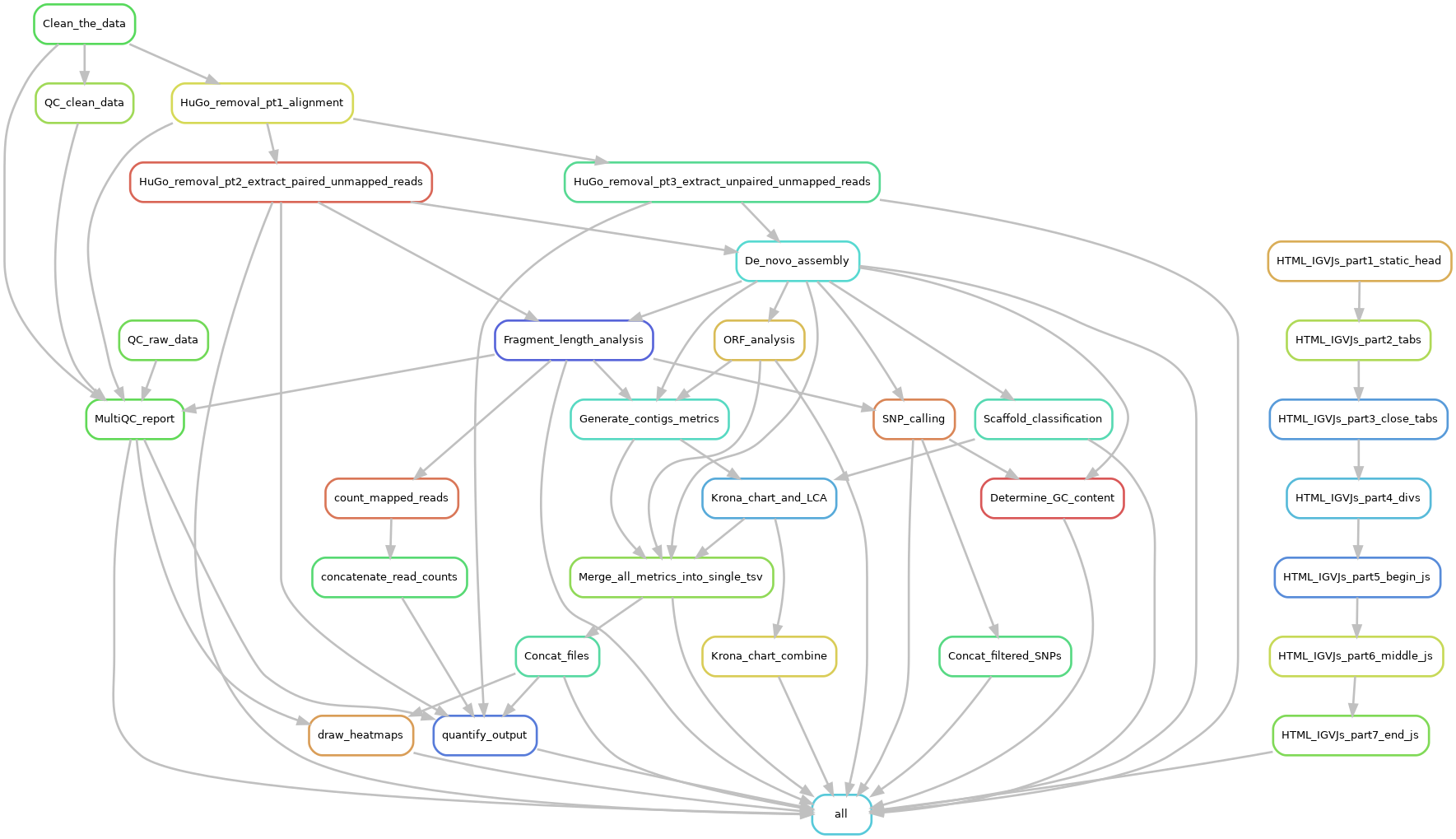
The pipeline automatically processes raw Illumina NGS data from human clinical matrices (faeces, serum, etc.) into clinically relevant information such as taxonomic classification, viral typing and minority variant identification (quasispecies). Wetlab personnel can start, configure and interpret results via an interactive web-report. This makes doing metagenomics analyses much more accessible and user-friendly since minimal command-line skills are required.
- Data quality control (QC) and cleaning.
- Including library fragment length analysis, useful for sample preparation QC.
- Removal of human* data (patient privacy). *You can use whichever reference you would like. However, Jovian is intended for human clinical samples.
- Assembly of short reads into bigger scaffolds (often full viral genomes).
- Taxonomic classification:
- Every nucleic acid containing biological entity (i.e. not only viruses) is determined up to species level.
- Lowest Common Ancestor (LCA) analysis is performed to move ambiguous results up to their last common ancestor, which makes results more robust.
- Viral typing:
- Several viral families and genera can be taxonomically labelled at the sub-species level as described here.
- Viral scaffolds are cross-referenced against the Virus-Host interaction database and NCBI host database.
- Scaffolds are annotated with great detail:
- Depth of coverage.
- GC content.
- Open reading frames (ORFs) are predicted.
- Minority variants (quasispecies) are identified.
- Importantly, results of all processes listed above are presented via an interactive web-report including an audit trail.
All data are visualized via an interactive web-report, as shown here, which includes:
- A collation of interactive QC graphs via
MultiQC. - Taxonomic results are presented on three levels:
- For an entire (multi sample) run, interactive heatmaps are made for non-phage viruses, phages and bacteria. They are stratified to different taxonomic levels.
- For a sample level overview,
Kronainteractive taxonomic piecharts are generated. - For more detailed analyses, interactive tables are included. Similar to popular spreadsheet applications (e.g. Microsoft Excel).
- Classified scaffolds
- Unclassified scaffolds (i.e. "Dark Matter")
- Virus typing results are presented via interactive spreadsheet-like tables.
- An interactive scaffold alignment viewer (
IGVjs) is included, containing:- Detailed alignment information.
- Depth of coverage graph.
- GC content graph.
- Predicted open reading frames (ORFs).
- Identified minority variants (quasispecies).
- All SNP metrics are presented via interactive spreadsheet-like tables, allowing detailed analysis.
After a Jovian analysis is finished you can perform virus-typing (i.e. sub-species level taxonomic labelling). These analyses can be started by the command bash jovian -vt [virus keyword], where [virus keyword] can be:
| Keyword | Taxon used for scaffold selection | Notable virus species |
|---|---|---|
NoV |
Caliciviridae | Norovirus GI and GII, Sapovirus |
EV |
Picornaviridae | Enteroviruses (Coxsackie, Polio, Rhino, etc.), Parecho, Aichi, Hepatitis A |
RVA |
Rotavirus A | Rotavirus A |
HAV |
Hepatovirus A | Hepatitis A |
HEV |
Orthohepevirus A | Hepatitis E |
PV |
Papillomaviridae | Human Papillomavirus |
Flavi |
Flaviviridae | Dengue (work in progress) |
An audit trail, used for clinical reproducability and logging, is generated and contains:
- A unique methodological fingerprint: allowing to exactly reproduce the analysis, even retrospectively by reverting to old versions of the pipeline code.
- The following information is also logged:
- Database timestamps
- (user-specified) Pipeline parameters
However, it has limitations since several things are out-of-scope for Jovian to control:
- The
virus typing-toolsversion- Currently we depend on a public web-tool hosted by the RIVM. These are developed in close collaboration with - but independently of - Jovian. A versioning system for the
virus typing-toolsis being worked on, however, this is not trivial and will take some time.
- Currently we depend on a public web-tool hosted by the RIVM. These are developed in close collaboration with - but independently of - Jovian. A versioning system for the
- Input files and metadata
- We only save the names and location of input files at the time the analysis was performed. Long-term storage of the data, and documenting their location over time, is the responsibility of the end-user. Likewise, the end-user is responsible for storing datasets with their correct metadata (e.g. clinical information, database versions, etc.). We recommend using iRODS for this as described by Nieroda et al. 2019. While we acknowledge that database versions are vital to replicate results, the databases Jovian uses have no official versioning, hence why we include timestamps only.
Can be found on this wiki page.
Below you'll find instructions on how to install and start/configure a Jovian analysis.
Can be found on this wiki page.
There are two methods, the first is by using the "Jovian Portal" and the second is via the command-line interface. The former is an interactive website that is intended for non-bioinformaticians while the latter is intended for people familiar with the command-line interface.
Jovian Portal:
- First, start a Jupyter notebook background process as described here. Or ask your system-admin to do this for you.
- Via the Jupyter Notebook connection established in the previous step, go to the
Jovianfolder created during installation. Then, openNotebook_portal.ipynb. - Follow the instructions in this notebook to start an analysis.
- N.B. These reports work with Mozilla Firefox and Google Chrome, we do not recommend using Internet Explorer.
Command-line interface:
- Make sure that Jovian is completely installed.
- Go to the folder where
Jovianwas installed. - Configure pipeline parameters by changing the profile/pipeline_parameters.yaml file. Either via Jupyter Notebook or with a commandline text-editor of choice.
- Optional: We recommended you do a
dry-runbefore each analysis to check if there are any typo's, missing files or other errors. This can be done viabash jovian -i <input_directory> -n - If the dry-run has completed without errors, you are ready to start a real analysis with the following command:
bash jovian -i <input_directory> - After analysis is finished, you can optionally genotype the viruses as described here (i.e. sub-species level taxonomic classification). This can be done via
bash jovian -vt [NoV|EV|RVA|HAV|HEV|PV|Flavi]. - After the pipeline has finished, open
Notebook_report.ipynbvia your browser (Mozilla Firefox or Google Chrome). Click onCellin the toolbar, then pressRun alland wait for data to be imported.- N.B. You need to have a Jupyter notebook process running in the background, as described here.
| Folder | Contents |
|---|---|
bin/ |
Contains the scripts required for Jovian to work |
data/ |
Contains intermediate and detailed data |
envs/ |
Contains all conda environment recipes for the pipeline |
files/ |
Contains ancillary files for the pipeline |
logs/ |
Contains all Jovian log files, use these files to troubleshoot errors |
profile/ |
Contains the files with Snakemake and pipeline parameters |
results/ |
This contains all files that are important for end-users and are imported by the Jupyter Report |
Also, a hidden folder named .snakemake is generated. Do not remove or edit this folder. Jovian was built via Snakemake and this folder contains all the software and file-metadata required for proper pipeline functionality.
Can be found on this wiki page.
Can be found on this wiki page.
| Name | Publication | Website |
|---|---|---|
| BBtools | NA | https://jgi.doe.gov/data-and-tools/bbtools/ |
| BEDtools | Quinlan, A.R. and I.M.J.B. Hall, BEDTools: a flexible suite of utilities for comparing genomic features. 2010. 26(6): p. 841-842. | https://bedtools.readthedocs.io/en/latest/ |
| BLAST | Altschul, S.F., et al., Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. 1997. 25(17): p. 3389-3402. | https://www.ncbi.nlm.nih.gov/books/NBK279690/ |
| BWA | Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997. | https://github.com/lh3/bwa |
| BioConda | Grüning, B., et al., Bioconda: sustainable and comprehensive software distribution for the life sciences. 2018. 15(7): p. 475. | https://bioconda.github.io/ |
| Biopython | Cock, P. J., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., ... & De Hoon, M. J. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25(11), 1422-1423. | https://biopython.org/ |
| Bokeh | Bokeh Development Team (2018). Bokeh: Python library for interactive visualization. | https://bokeh.pydata.org/en/latest/ |
| Bowtie2 | Langmead, B. and S.L.J.N.m. Salzberg, Fast gapped-read alignment with Bowtie 2. 2012. 9(4): p. 357. | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Conda | NA | https://conda.io/ |
| DRMAA | NA | http://drmaa-python.github.io/ |
| FastQC | Andrews, S., FastQC: a quality control tool for high throughput sequence data. 2010. | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| gawk | NA | https://www.gnu.org/software/gawk/ |
| GNU Parallel | O. Tange (2018): GNU Parallel 2018, March 2018, https://doi.org/10.5281/zenodo.1146014. | https://www.gnu.org/software/parallel/ |
| Git | NA | https://git-scm.com/ |
| igvtools | NA | https://software.broadinstitute.org/software/igv/igvtools |
| Jupyter Notebook | Kluyver, Thomas, et al. "Jupyter Notebooks-a publishing format for reproducible computational workflows." ELPUB. 2016. | https://jupyter.org/ |
| Jupyter_contrib_nbextension | NA | https://github.com/ipython-contrib/jupyter_contrib_nbextensions |
| Jupyterthemes | NA | https://github.com/dunovank/jupyter-themes |
| Krona | Ondov, B.D., N.H. Bergman, and A.M. Phillippy, Interactive metagenomic visualization in a Web browser. BMC Bioinformatics, 2011. 12: p. 385. | https://github.com/marbl/Krona/wiki |
| Lofreq | Wilm, A., et al., LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. 2012. 40(22): p. 11189-11201. | http://csb5.github.io/lofreq/ |
| MGkit | Rubino, F. and Creevey, C.J. 2014. MGkit: Metagenomic Framework For The Study Of Microbial Communities. . Available at: figshare [doi:10.6084/m9.figshare.1269288]. | https://bitbucket.org/setsuna80/mgkit/src/develop/ |
| Minimap2 | Li, H., Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 2018. | https://github.com/lh3/minimap2 |
| MultiQC | Ewels, P., et al., MultiQC: summarize analysis results for multiple tools and samples in a single report. 2016. 32(19): p. 3047-3048. | https://multiqc.info/ |
| Nb_conda | NA | https://github.com/Anaconda-Platform/nb_conda |
| Nb_conda_kernels | NA | https://github.com/Anaconda-Platform/nb_conda_kernels |
| Nginx | NA | https://www.nginx.com/ |
| Numpy | Walt, S. V. D., Colbert, S. C., & Varoquaux, G. (2011). The NumPy array: a structure for efficient numerical computation. Computing in Science & Engineering, 13(2), 22-30. | http://www.numpy.org/ |
| Pandas | McKinney, W. Data structures for statistical computing in python. in Proceedings of the 9th Python in Science Conference. 2010. Austin, TX. | https://pandas.pydata.org/ |
| Picard | NA | https://broadinstitute.github.io/picard/ |
| Prodigal | Hyatt, D., et al., Prodigal: prokaryotic gene recognition and translation initiation site identification. 2010. 11(1): p. 119. | https://github.com/hyattpd/Prodigal/wiki/Introduction |
| Python | G. van Rossum, Python tutorial, Technical Report CS-R9526, Centrum voor Wiskunde en Informatica (CWI), Amsterdam, May 1995. | https://www.python.org/ |
| Qgrid | NA | https://github.com/quantopian/qgrid |
| SAMtools | Li, H., et al., The sequence alignment/map format and SAMtools. 2009. 25(16): p. 2078-2079. | http://www.htslib.org/ |
| SPAdes | Nurk, S., et al., metaSPAdes: a new versatile metagenomic assembler. Genome Res, 2017. 27(5): p. 824-834. | http://cab.spbu.ru/software/spades/ |
| Seqtk | NA | https://github.com/lh3/seqtk |
| Snakemake | Köster, J. and S.J.B. Rahmann, Snakemake—a scalable bioinformatics workflow engine. 2012. 28(19): p. 2520-2522. | https://snakemake.readthedocs.io/en/stable/ |
| Tabix | NA | www.htslib.org/doc/tabix.html |
| tree | NA | http://mama.indstate.edu/users/ice/tree/ |
| Trimmomatic | Bolger, A.M., M. Lohse, and B. Usadel, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 2014. 30(15): p. 2114-20. | www.usadellab.org/cms/?page=trimmomatic |
| Virus-Host Database | Mihara, T., Nishimura, Y., Shimizu, Y., Nishiyama, H., Yoshikawa, G., Uehara, H., ... & Ogata, H. (2016). Linking virus genomes with host taxonomy. Viruses, 8(3), 66. | http://www.genome.jp/virushostdb/note.html |
| Virus typing tools | Kroneman, A., Vennema, H., Deforche, K., Avoort, H. V. D., Penaranda, S., Oberste, M. S., ... & Koopmans, M. (2011). An automated genotyping tool for enteroviruses and noroviruses. Journal of Clinical Virology, 51(2), 121-125. | https://www.ncbi.nlm.nih.gov/pubmed/21514213 |
- Dennis Schmitz (RIVM and EMC)
- Sam Nooij (RIVM and EMC)
- Robert Verhagen (RIVM)
- Thierry Janssens (RIVM)
- Jeroen Cremer (RIVM)
- Florian Zwagemaker (RIVM)
- Mark Kroon (RIVM)
- Erwin van Wieringen (RIVM)
- Harry Vennema (RIVM)
- Annelies Kroneman (RIVM)
- Marion Koopmans (EMC)
This project/research has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 643476.