#ContaVect
Contavect is a python2.7 object oriented script, developed to quantify and characterize DNA contaminants from gene therapy vector production after NGS sequencing. This automated pipeline can however be used for wider purpose requiring to identify map NGS datasets consisting of a mix of DNA sequences on multiple references. It combine several features such as reference homologies masking, fastq filtering/adapter trimming, short read alignments, SAM file splitting and generating human readable output.
##Principle
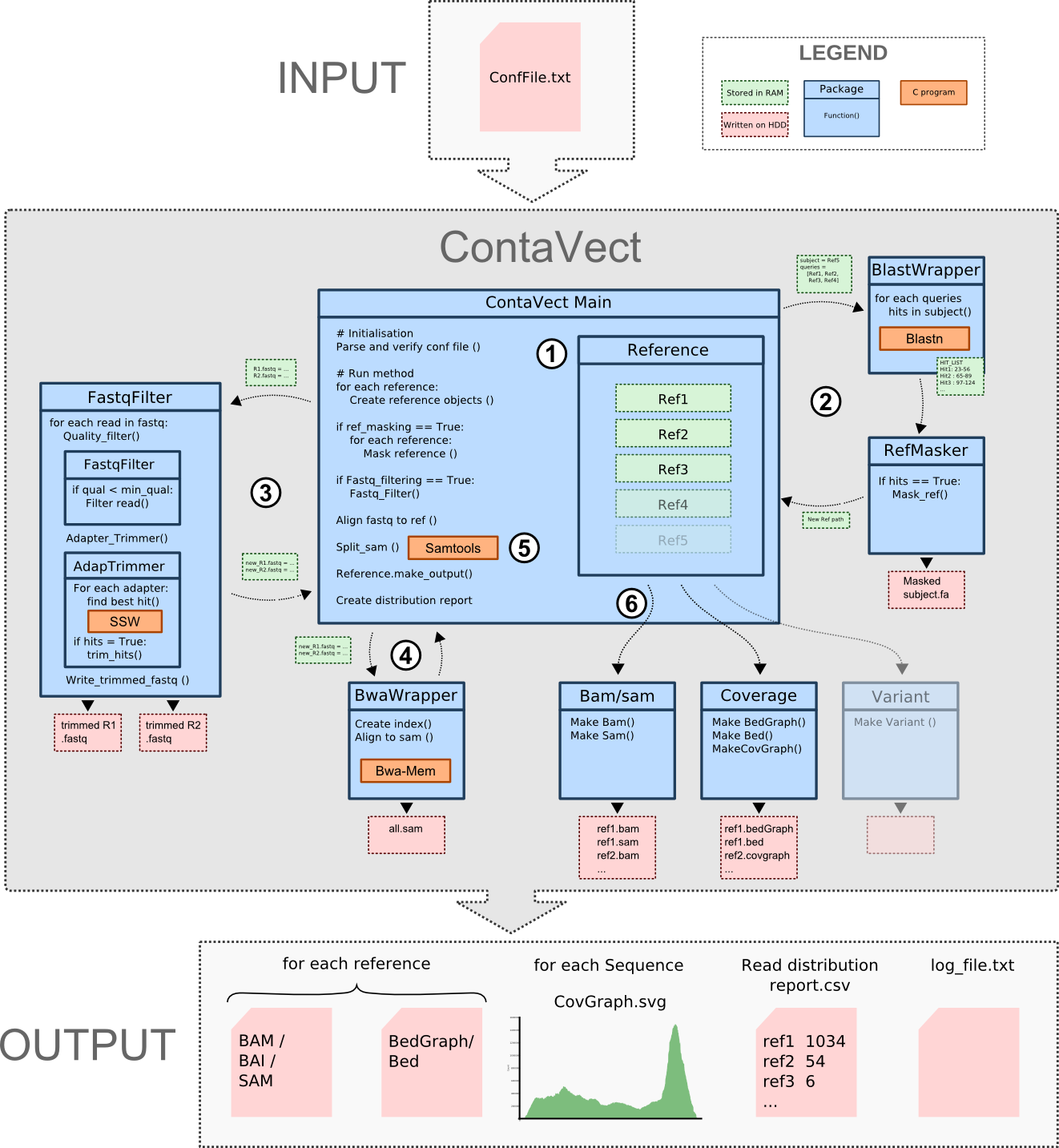
Contavect a python pipeline composed of several modules linked together to analyze NGS Data. Here is a description of the overall workflow principle :
- Each reference fasta file is parsed to identify all sequences within it and a Reference object is initialized to save the reference characteristics, the name and the output required.
- Facultative: Homologies between references can be masked iteratively, starting by the last reference which is masked by all the others then to the penultimate masked by all others except the last and and so forth until there is only 1 reference remaining. This is done using blastn from blast+ package
- Facultative: Fastq can be filtered by mean quality and adapters can be trimmed using an homemade fully integrated fastq filter parallel processing module written in python and C.
- If needed an index for bwa will be generated from the modified reference files or from the original one after being merged together in a temporary directory. Then Fastq sequences are then aligned against the bwa merged reference genome index and a temporary sam file is generated
- Aligned reads from the sam file are spitted and attributed to the reference Object for which a hit was found. or to one of the following garbage reads categories: unmaped, lowMapq, secondary.
- Each reference will then generates the output required in the configuration file (Bam, sam, bedgraph, bed and covgraph).
- Finally distribution reports and a log file are generated
For more information, a comprehensive developer documentation can be generated from ContaVect.dox using Doxygen with doxypy.
The program was developed under Linux Mint 16/17 and require a python 2.7 environment. The following dependencies are required for proper program execution:
- bwa 0.7.0+
- samtools 0.1.17+
- blast+ (not required is not no reference masking is to be performed)
In addtion 2 third party python packages are also needed
If you have pip already installed, enter the following line to install pysam:
sudo pip install pysam- Clone the repository with --recursive option to also pull the submodule
$ git clone --recursive https://github.com/a-slide/ContaVect/ my_folder/- Enter the root of the program folder and make the main script executable
$ sudo chmod u+x ContaVect.py3.Compile the ssw aligner (and add the dynamic library it to the path)
If you wish to perform a step of adapter trimming before mapping you need to complile the dynamic library ssw.so to be able to use the Smith Waterman algorithm forked from mengyao's Complete-Striped-Smith-Waterman-Library
To use the dynamic library libssw.so you may need to modify the LD_LIBRARY_PATH environment variable to include the library directory (export LD_LIBRARY_PATH=$PWD) or for definitive inclusion of the lib edit /etc/ld.so.conf and add the path or the directory containing the library and update the cache by using /sbin/ldconfig as root
Prepare the configuration file to include your files and settings as indicated in the template Conf.txt file provided with the source files
$ ./ContaVect.py Conf.txt No command line option is available, everything is in the Configuration file
Export the path to conf/matplotlibrc file as the MATPLOTLIBRC variable for headless cluster runs.
2 possibilities:
- Use ipython notebook with doc/Logbook.ipynb
- Consult directly online through nbviewer : Notebook
- Adrien Leger aleg@ebi.ac.uk @a-slide
- Emilie Lecomte emilie.lecomte@univ-nantes.fr @emlec