Kenya Virome
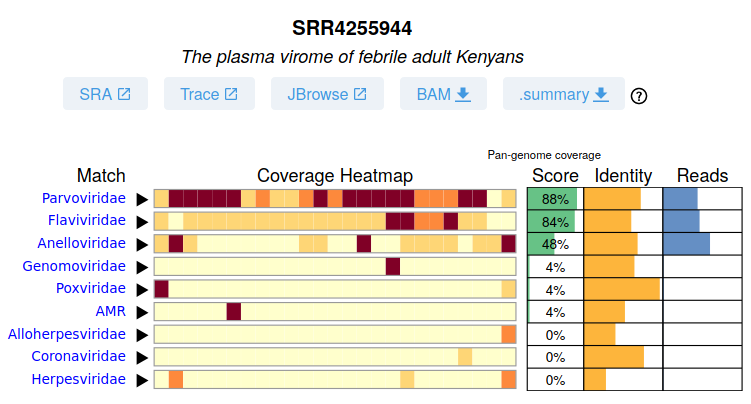
It should be immediatly clear this samples contains, with high confidence, a Parvovirus and a Flavivirus. With moderate confidence there is an Anellovirus.
Note that for these three viral pangenomes there many reads (438, 463, and 595) spread across several 'bins' as shown in the heatmap, an indicator for a real hit.
In contrast there are 7 reads matching a Genomovirius, all in a single bin which is more likely to be a spurious match.
Clicking on the "Coverage Heatmap" expands to show the top 'sequences' in this pangenome with aligned reads. Here it is NC_001710.1 Hepatitis G virus, and NC_001474.2 Dengue virus 2.
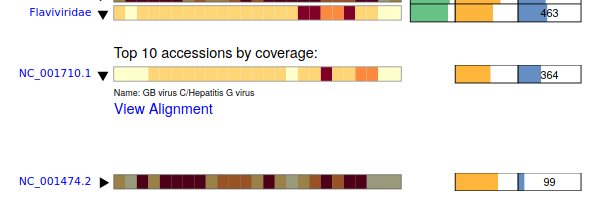
Further expanding Hepatitis G shows there are 364 reads at an average nucleotide identity of 89.5%. A critical note here is that the nucleotide identity corresponds only to the 'mapped' portion of the reads which can be 'soft clipped'. Also nucleotide reads are most likely to map to 'conserved' regions of the genomes such as polymerases and thus over-estimate true genome-wide nucleotide identity.
To validate if this is infact a hepatitis G virus, click "View Alignment" to open a JBrowse session for this SRA/Virus.
JBrowse Session for SRR4255944
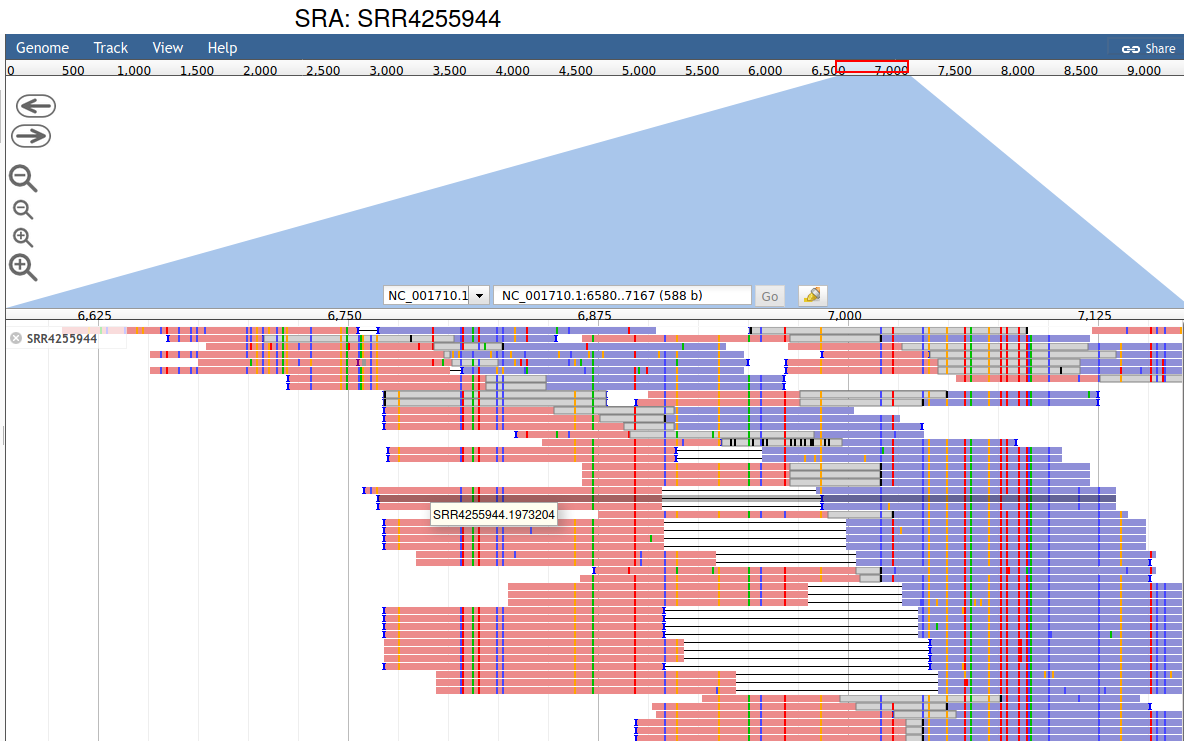
Exploring the aligned reads it's clear there are quite a few variations present at the nucleotide level, and the coverage is uneven, this is expected for sequences 'diverged' from the RefSeq reference.
Clicking on a read will open "read details"
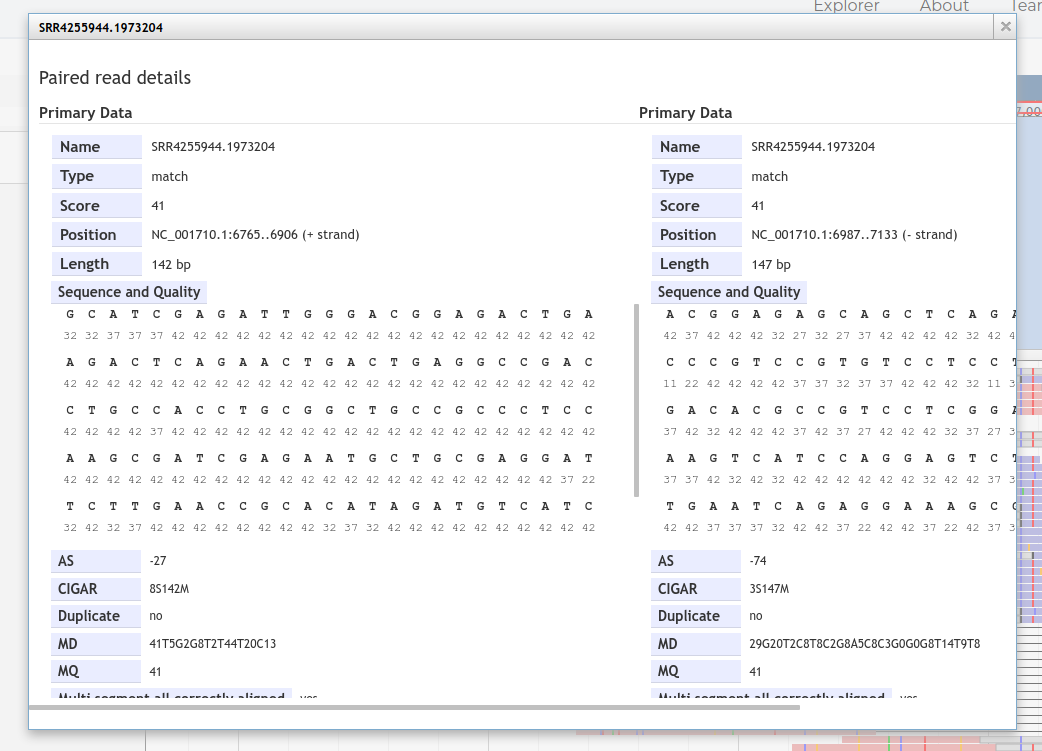
To confirm this is 'true positive' and not a 'false positive' spurious match, copy the read sequence and perform both a blastn and blastx search of the read.
blastn
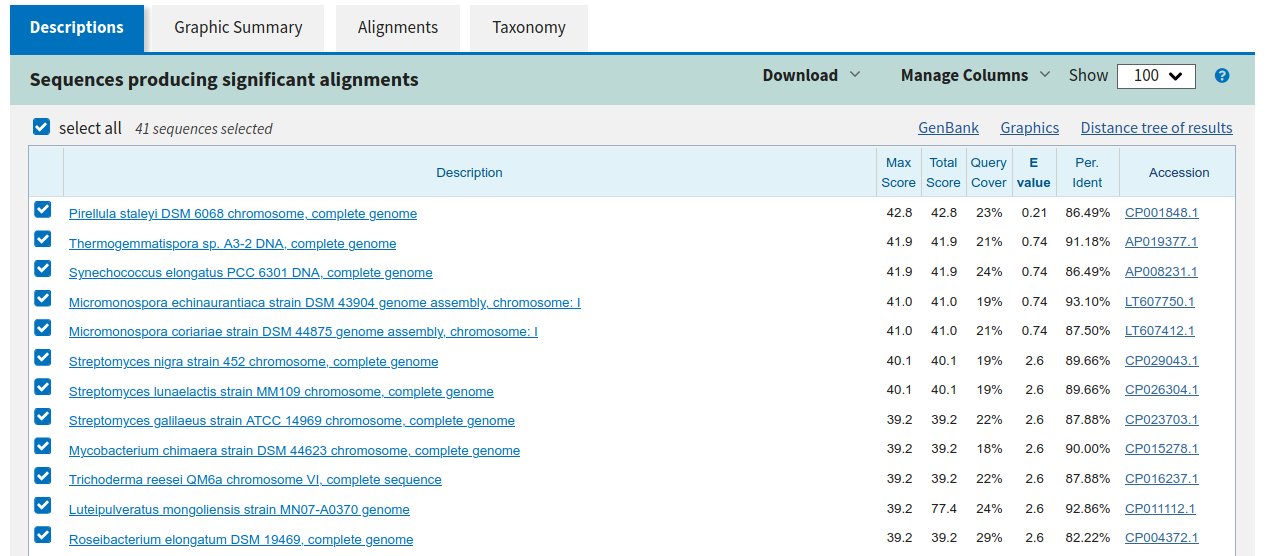
Note the match only covers a small region of the query (23%) and the E-value (expectence) is quite high therefore not confident.
blastx
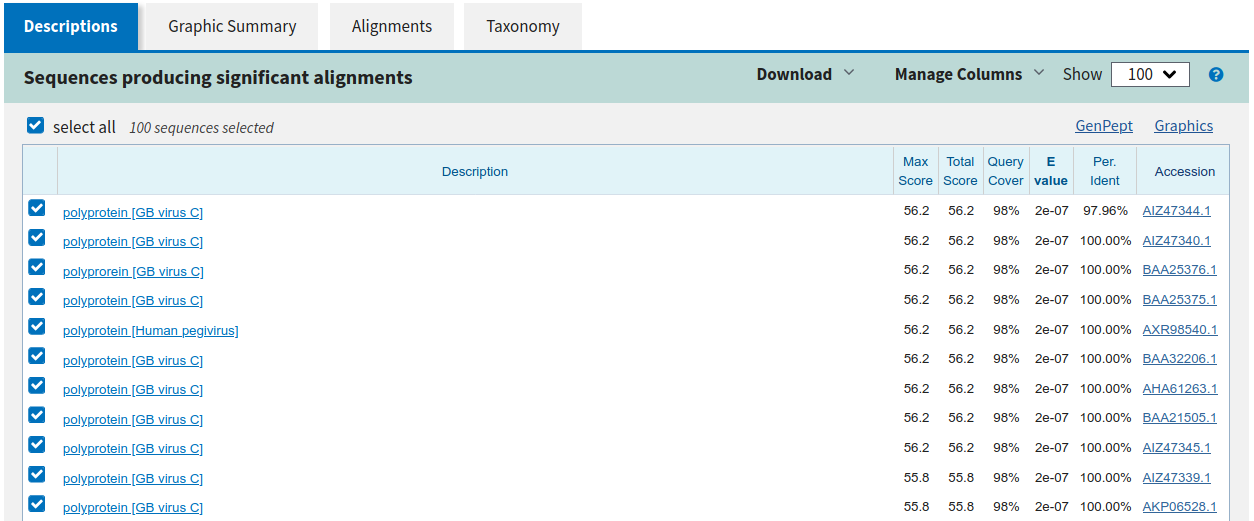
At the protein level, one can 'align' more deeply and here you can see very high identity for Heptatis G virus polyprotein, thus this is a true positive hit worth moving forward with de novo assembly.
The Score 48 Anellovirus is a moderate hit, coupled with a lower sequence idenity (92%) makes this a prime candidate for a sequence diverse virus.
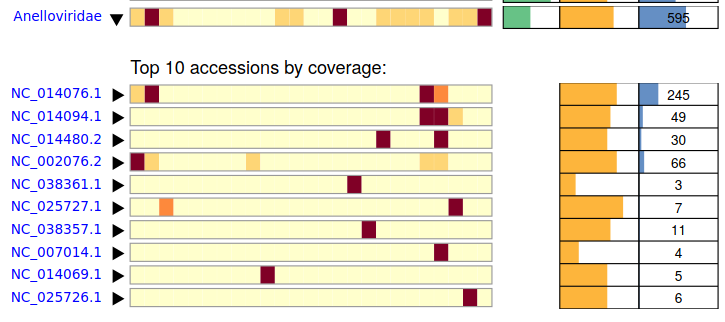
As before, view the alignment directly to make a judgement if this is a true positive hit.

There are two regions of sequence coverage; the 5' reads span only 120 nt strictly overlapping; the 3' reads span 318 nt with read-pairs in the same region of the genone which is a good sign.
As before, blastn and blastx reads from each cluster:
5' blastx
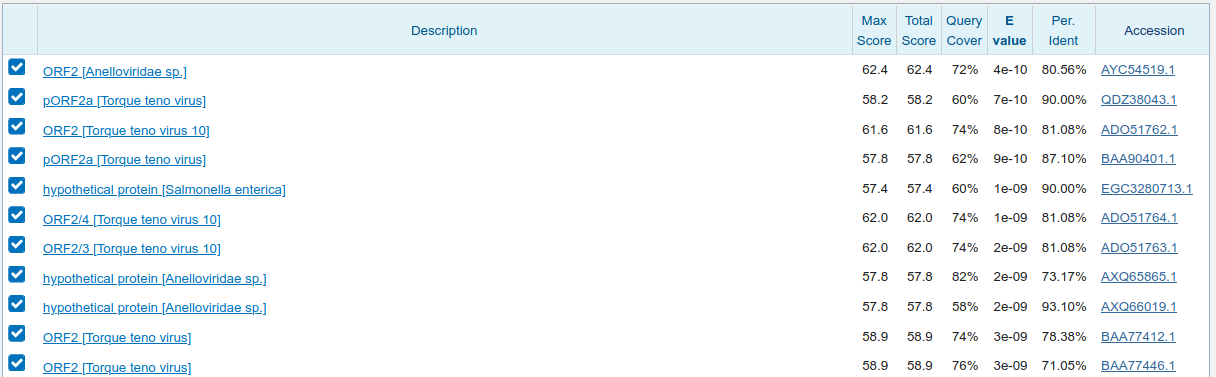
3' blastx
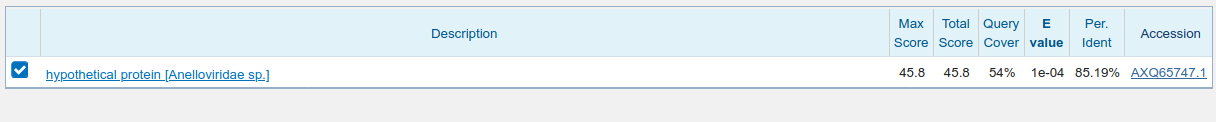
The translated sequences both match Anelloviridae in GenBank at ~80% protein identity from an 'aligned' nucleotide identity of 93%. This would therefore be a high priority for de novo assembly as the library contains what is likely an uncharacterized human Anellovirus.
At this point it's worth tracing back the read to it's bioProject PRJNA343480.
One you have identified a 'match pattern', it is possible to re-query the Serratus data and find similar matches.
For the Anellovirus example above, we identified a virus with 93% identity to NC_014076.1 and a coverage of 16% (4/25 bins). Therefore a "GenBank" search for 90-95% identity and 4-25% coverage is likely to yield more similar viruses.