Binning

fig: The Genious document table, showing the bin field, and other useful quality measures
"Binning" is used to group contigs and reads into three categories (High, Medium and Low) based on various measures of quality (see Binning Parameters below). It's purpose is to speed up processing by summarizing the properties of sequencing results which the researcher would normally have to sift through manually.
The Bin column is automatically added to all sequences and contigs in Geneious. However, the Bin column is hidden by default and needs to be turned on first. To do this, click on the small icon in the top-right corner of the table then check the Bin item. This can also be done by right clicking on the table header.
Documents can be sorted according to Bin by clicking on the Bin table header (see figure).
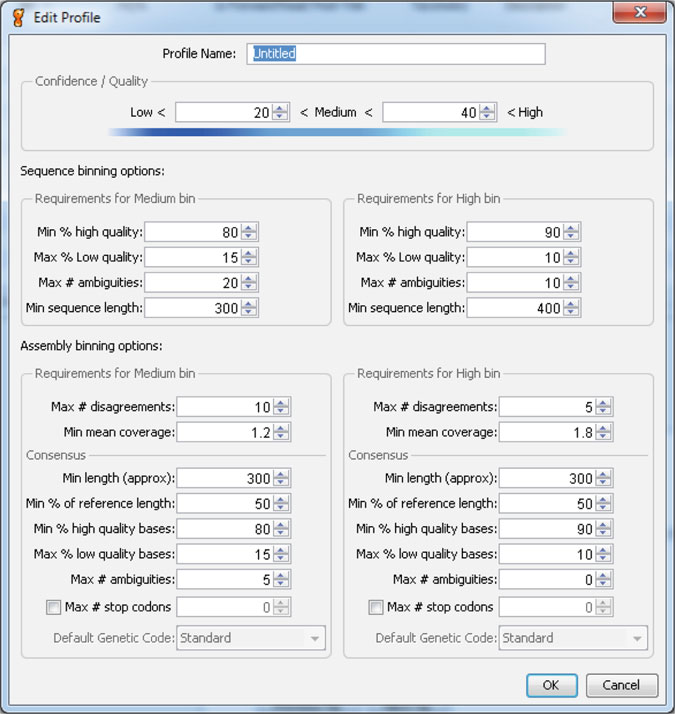
fig: The Binning Parameters Dialog
The binning parameters tell Geneious what the cut off points are for each of the bins. The binning parameters cover metrics such as the percentages of high and low quality bases in your sequences, sequence length, number of ambiguities, and coverage in the case of assemblies. To check the full list of parameters, see the binning parameters dialog (screenshot to the right). For information on any of the binning parameters, hold the mouse over the option to get a description.
There are three levels at which binning parameters can be set: globally (for all local and server documents), per-folder (all documents inside a particular folder or any subfolder) and per-document. To set the global binning parameters, select Tools > Preferences... from the menu and go to the Sequencing tab. To set per-folder or per-document parameters, select the folder or documents you want to change then go to Sequence > Set Binning Parameters... in the menu.
The most specific parameters are used in favor of less specific ones. So per-document parameters will be used over any per-folder or global parameters that are set.
One optional binning parameter for assemblies is the number of stop codons. The number of stop codons is calculated for the specified genetic code, and is defined as the minimum count of stop codons in the consensus sequence for all frames (ie we check frames 1, 2, 3 in the forward and reverse direction, count the number of stop codons in each, and then take frame with the minimum number of stops). Setting the maximum of this value to 0 for the high bin ensures that you do not have frameshifts in your sequences, and instantly draws your attention to trouble data.
When looking at assembly results, the bins could be used in the following way (if the parameters as set up as such):
- High = Perfect! No need to look at these contigs
- Medium = Suspect. Look at these contigs to make sure they are alright (see Viewing and Editing Contigs section)
- Low = Fail. These are beyond rescue so mark them for resequencing and investigation in to what went wrong
Mean coverage is one of the binning criteria for contigs and is also available as a column in the table. It is also the least intuitive value so here is a description:
Coverage is the number of sequences that cover a given position in an alignment/contig. Mean coverage is therefore the mean of this value across all positions in the alignment/contig.

For this alignment the first two positions have a coverage of 1. The next five positions have a coverage of 2 and the last three have coverage 1 again. Mean coverage is therefore (2*1 + 5*2 + 3*1) / 10 = 1.5
The mean coverage will be between 1 and the number of sequences in the alignment/contig. For a pairwise assembly that means 2 is full coverage and 1 is no coverage.
To set up the parameters in this way you will need to have strict parameters for the high bin, perhaps 0 disagreements, 650 consensus length and 1.8 coverage (for the COI barcode). The medium bin can be quite relaxed depending on how many contigs you want to examine.