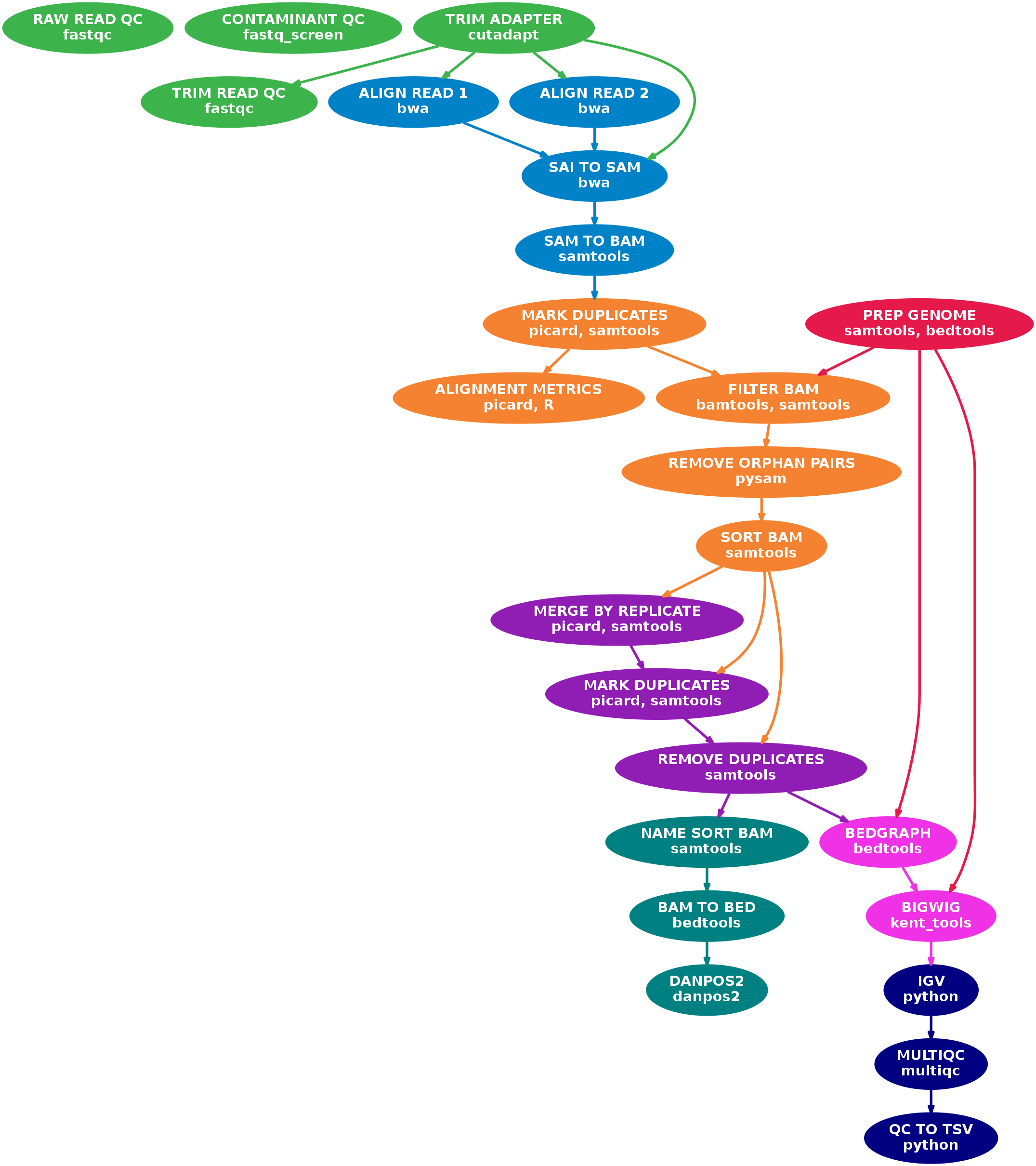
A Nextflow pipeline for processing paired-end Illumina MNASeq sequencing data.
The pipeline was written by The Bioinformatics & Biostatistics Group at The Francis Crick Institute, London.
- Raw read QC (
FastQC,Fastq Screen) - Adapter trimming (
cutadapt) - Alignment (
BWA) - Mark duplicates (
picard) - Filtering to remove:
- reads that are marked as duplicates (
SAMtools) - reads that arent marked as primary alignments (
SAMtools) - reads that are unmapped (
SAMtools) - reads that map to multiple locations (
SAMtools) - reads containing > 3 mismatches in either read of the pair (
BAMTools) - reads that have a user-defined insert size (
BAMTools) - reads that are soft-clipped (
BAMTools) - reads that map to different chromosomes (
Pysam) - reads that arent in FR orientation (
Pysam) - reads where only one read of the pair fails the above criteria (
Pysam)
- reads that are marked as duplicates (
- Merge alignments at replicate-level (
picard)- Re-mark duplicates (
picard) - Remove duplicate reads (optional;
SAMtools) - Create normalised bigWig files scaled to 1 million mapped read pairs (
BEDTools,wigToBigWig)
- Re-mark duplicates (
- Call nucleosome positions and generate smoothed, normalised coverage wig files that can be used to generate occupancy profile plots between samples across features of interest (
DANPOS2) - Create IGV session file containing bigWig tracks for data visualisation (
IGV) - Collect and present QC at the raw read and alignment-level (
MultiQC)
The documentation for the pipeline can be found in the docs/ directory:
- Installation
- Pipeline configuration
- Reference genome
- Design file
- Running the pipeline
- Output and interpretation of results
- Troubleshooting
The pipeline was written by the The Bioinformatics & Biostatistics Group at The Francis Crick Institute, London.
The pipeline was developed by Harshil Patel.
The NGI-RNAseq pipeline developed by Phil Ewels was used a template for this pipeline. Many thanks to Phil and the team at SciLifeLab.
This project is licensed under the MIT License - see the LICENSE.md file for details.