


SVIM-asm (pronounced SWIM-assem) is a structural variant caller for haploid or diploid genome-genome alignments. It analyzes a given sorted BAM file (preferably from minimap2) and detects five different variant classes between the query assembly and the reference: deletions, insertions, tandem and interspersed duplications and inversions.
Note! To analyze raw long sequencing reads please use our other method SVIM.
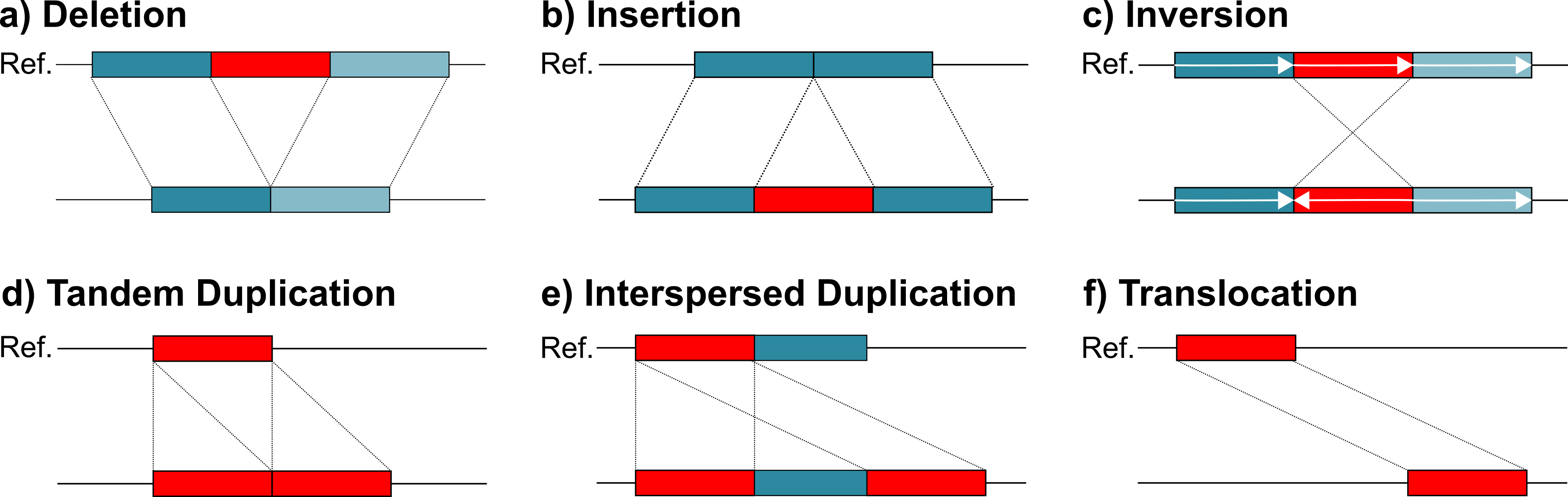
Structural variants (SVs) are typically defined as genomic variants larger than 50bps (e.g. deletions, duplications, inversions). Studies have shown that they affect more bases in an average genome than all SNPs or all small Indels together. Consequently, they have a large impact on genes and regulatory regions. This is reflected in the large number of genetic disorders and other disease that are associated to SVs.
Nowadays, SVs are usually detected using data from second-generation sequencing (Illumina) or third-generation sequencing (PacBio and Oxford Nanopore). Typically, the reads from a sequencing experiment are first aligned to a reference genome before the alignments are analyzed for characteristic signatures of SVs. Recently, substantial advances in sequencing technology and software development have made the de novo assembly of large mammalian genomes more efficient than ever. Accurate assemblies of the human genome can now be generated in a few days and at a fraction of its former cost. (Shafin et al.)
Similarly to raw sequencing reads, the genome assemblies can be aligned to another genome to uncover genomic rearrangements and structural variants. Our tool, SVIM-asm, detects structural variants between different assemblies or reference genomes from given genome-genome alignments. It is fast (<5 min for a human genome-genome alignment), easy to use and detects all major variant types.
SVIM-asm can be installed most easily using conda:
#Recommended: Install via conda into a new environment
conda create -n svimasm_env --channel bioconda svim-asm
#Alternatively: Install via conda into existing (active) environment
conda install --channel bioconda svim-asmAlternatively, SVIM-asm can be installed using pip:
#Install from github (requires Python 3)
git clone https://github.com/eldariont/svim-asm.git
cd svim-asm
pip install .- v1.0.3: bugfixes
- v1.0.2: change default value for partitioning, fix coordinates of BNDs and sorting of VCF records, add verbose mode
- v1.0.1: reduce memory consumption substantially
- v1.0.0: add genotyping of translocation breakpoints (BNDs), bugfixes
- v0.1.1: improve breakend detection, add FORMAT:CN tag for tandem duplications, add two new command-line options to output duplications as INS records in VCF, bugfixes
- v0.1.0: initial beta release
SVIM-asm analyzes alignments between a query assembly and a reference assembly in SAM/BAM format. We recommend to produce the alignments using minimap2. See this example for a haploid query assembly:
minimap2 -a -x asm5 --cs -r2k -t <num_threads> <reference.fa> <assembly.fasta> > <alignments.sam>
samtools sort -m4G -@4 -o <alignments.sorted.bam> <alignments.sam>
samtools index <alignments.sorted.bam>
svim-asm haploid <working_dir> <alignments.sorted.bam> <reference.fa>To analyze a diploid assembly consisting of two haplotypes, you need to align both to the reference assembly:
minimap2 -a -x asm5 --cs -r2k -t <num_threads> <reference.fa> <haplotype1.fasta> > <alignments_hap1.sam>
minimap2 -a -x asm5 --cs -r2k -t <num_threads> <reference.fa> <haplotype2.fasta> > <alignments_hap2.sam>
samtools sort -m4G -@4 -o <alignments_hap1.sorted.bam> <alignments_hap1.sam>
samtools sort -m4G -@4 -o <alignments_hap2.sorted.bam> <alignments_hap2.sam>
samtools index <alignments_hap1.sorted.bam
samtools index <alignments_hap2.sorted.bam
svim-asm diploid <working_dir> <alignments_hap1.sorted.bam> <alignments_hap2.sorted.bam> <reference.fa>SVIM-asm creates all output files in the given working directory. The following files are produced:
variants.vcfcontains the detected SVs in VCF format (see http://samtools.github.io/hts-specs/VCFv4.2.pdf)sv-lengths.pngcontains a histogram of SV sizesSVIM_<day>_<time>.logcontains the same logging output as the command line
If you experience problems or have suggestions please create an issue. As this project is not actively maintained anymore, however, we can not promise that they will be addressed in time.
Feel free to read and cite our paper in Bioinformatics: SVIM-asm: Structural variant detection from haploid and diploid genome assemblies
The project is licensed under the GNU General Public License.