

Simple FASTQ, SAM and BAM read quality assessment and plotting using Python.
- Requires only Python with Numpy, Scipy, and Matplotlib libraries
- Works with (gzipped) FASTQ, SAM, and BAM formatted reads
- Tabular, tidy, output statistics so you can create your own graphs
- A useful set of default graphics rivaling comparable QC packages
- Counts all IUPAC ambiguous nucleotide codes (NMWSKRYVHDB) if present in sequences
- Downsamples input files to around 2,000,000 reads (user adjustable)
- Allows a 5′ and 3′ (left and right) cycle limit for graphics generation
- Tracks kmers and sequence duplication for the entire input file
- Plots base call reference mismatches for aligned reads
- Optional sequence duplication calculation using Bloom filters (beta)
Tested on Python 2.7, and 3.4
Tested on Mac OS 10.10 and Linux 2.6.18
pip install [--user] fastqp
Note: BAM file support requires samtools
usage: fastqp [-h] [-q] [-s BINSIZE] [-a NAME] [-n NREADS] [-p BASE_PROBS] [-k {2,3,4,5,6,7}] [-o OUTPUT]
[-ll LEFTLIMIT] [-rl RIGHTLIMIT] [-mq MEDIAN_QUAL] [--aligned-only | --unaligned-only] [-d]
input
simple NGS read quality assessment using Python
positional arguments:
input input file (one of .sam, .bam, .fq, or .fastq(.gz) or stdin (-))
optional arguments:
-h, --help show this help message and exit
-q, --quiet do not print any messages (default: False)
-s BINSIZE, --binsize BINSIZE
number of reads to bin for sampling (default: auto)
-a NAME, --name NAME sample name identifier for text and graphics output (default: input file name)
-n NREADS, --nreads NREADS
number of reads sample from input (default: 2000000)
-p BASE_PROBS, --base-probs BASE_PROBS
probabilites for observing A,T,C,G,N in reads (default: 0.25,0.25,0.25,0.25,0.1)
-k {2,3,4,5,6,7}, --kmer {2,3,4,5,6,7}
length of kmer for over-repesented kmer counts (default: 5)
-o OUTPUT, --output OUTPUT
base name for output files (default: fastqp_figures)
-ll LEFTLIMIT, --leftlimit LEFTLIMIT
leftmost cycle limit (default: 1)
-rl RIGHTLIMIT, --rightlimit RIGHTLIMIT
rightmost cycle limit (-1 for none) (default: -1)
-mq MEDIAN_QUAL, --median-qual MEDIAN_QUAL
median quality threshold for failing QC (default: 30)
--aligned-only only aligned reads (default: False)
--unaligned-only only unaligned reads (default: False)
-d, --count-duplicates
calculate sequence duplication rate (default: False)
See releases page for details.
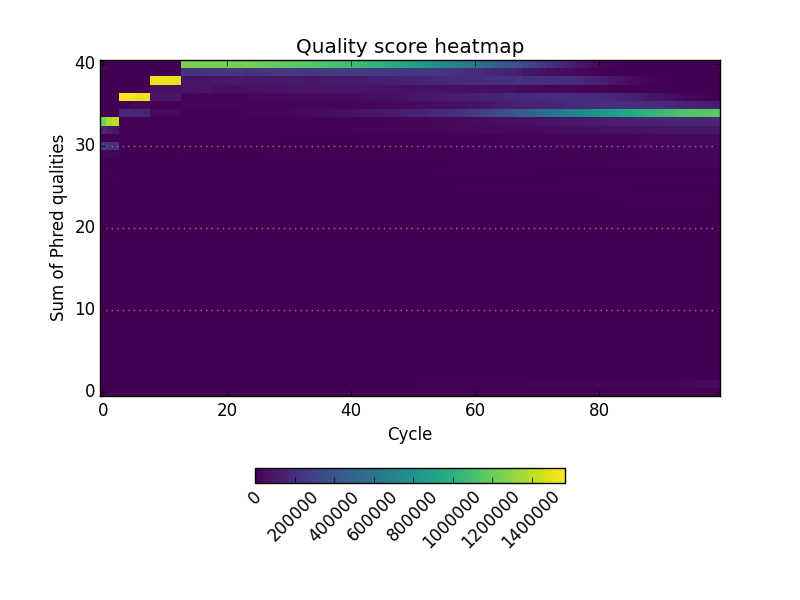
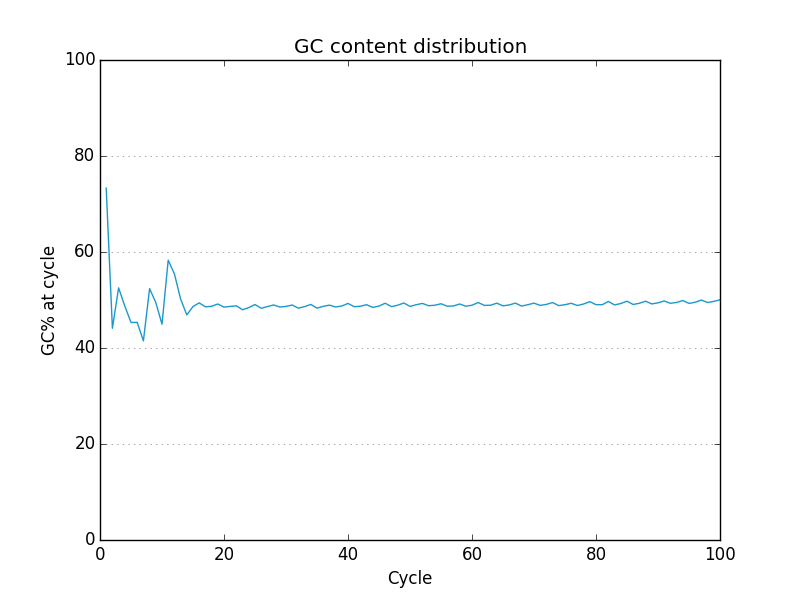
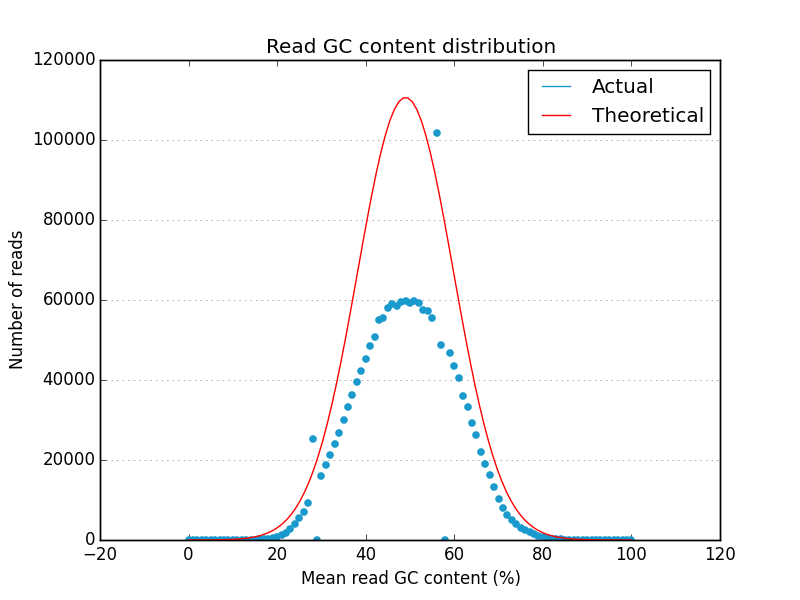
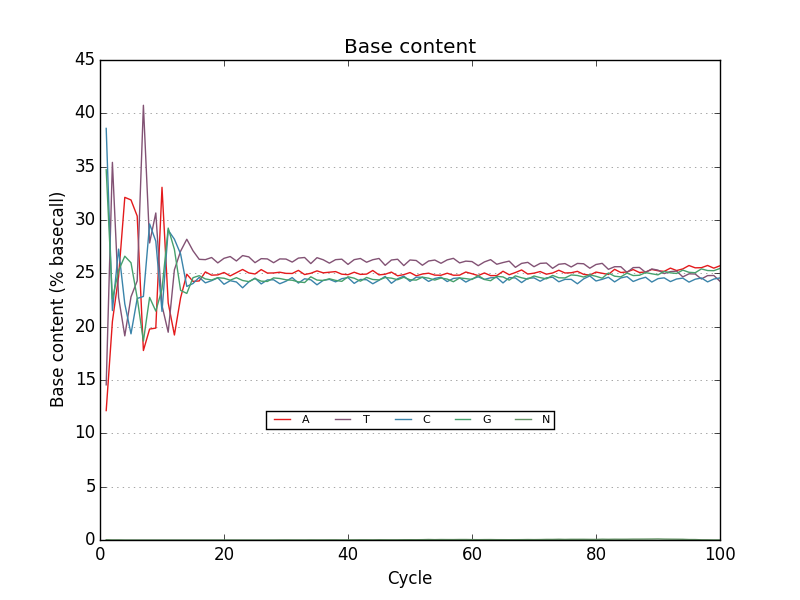
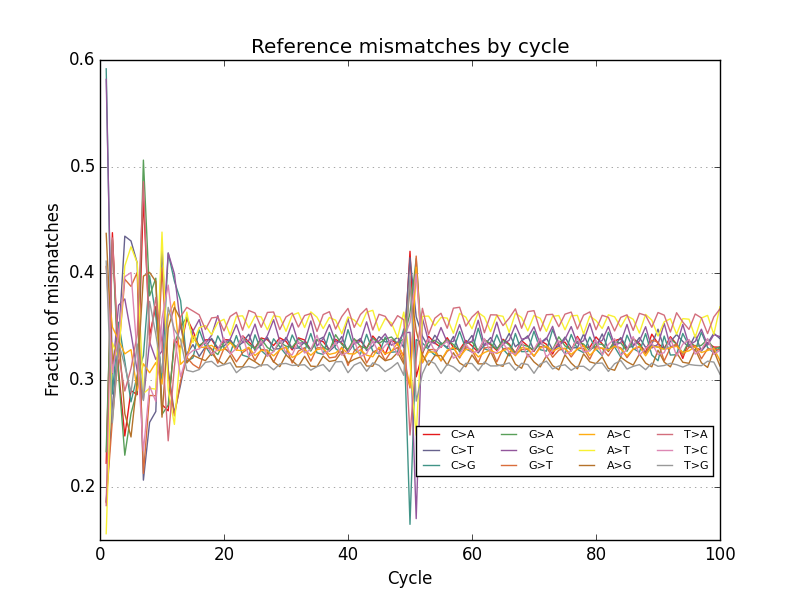
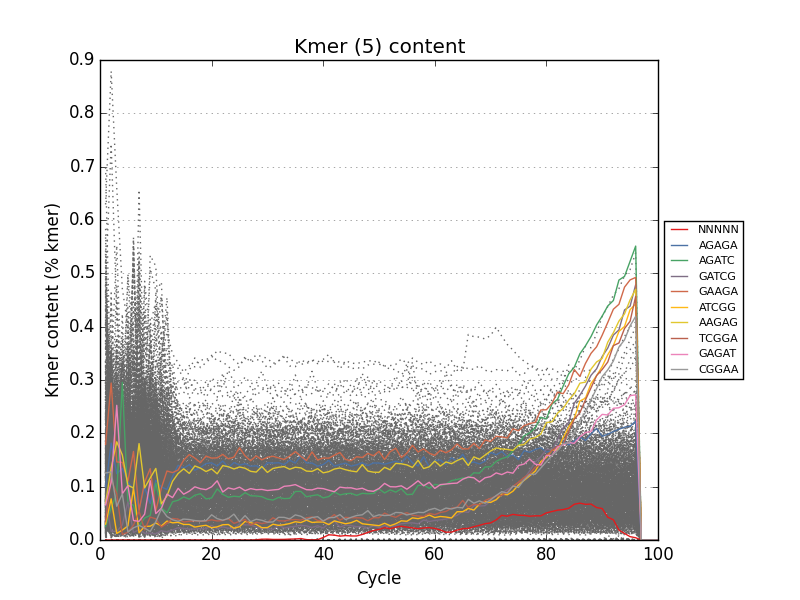
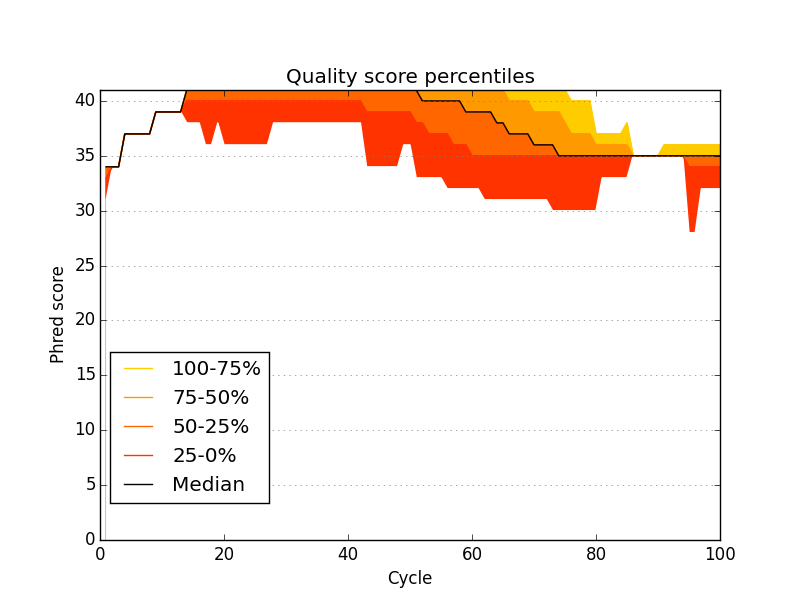
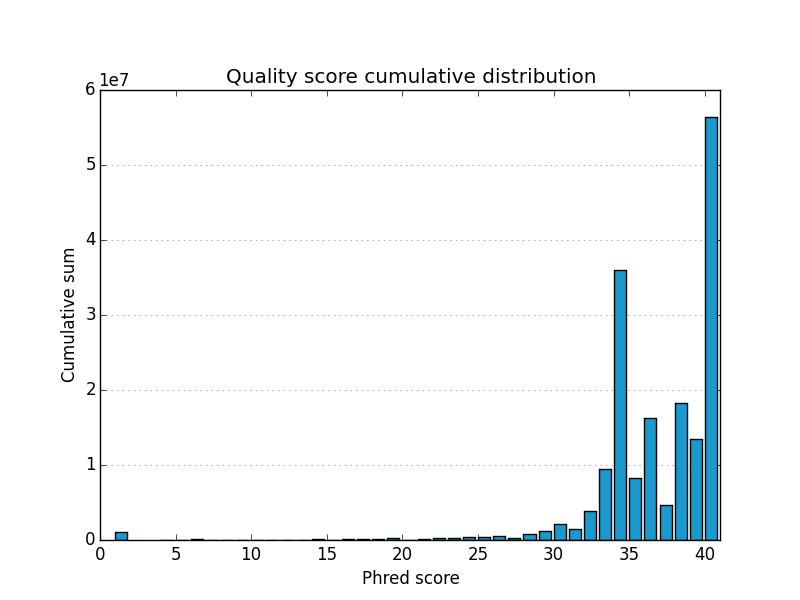
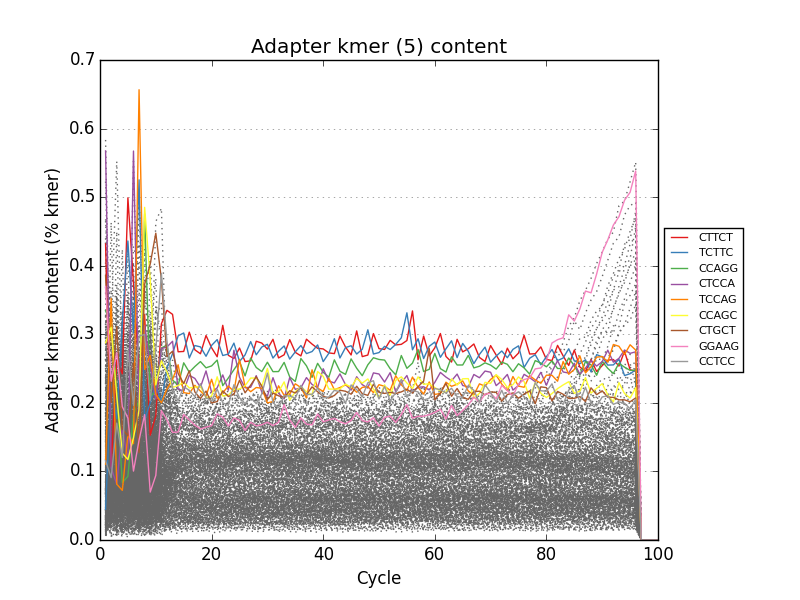
This project is freely licensed by the author, Matthew Shirley, and was completed under the mentorship financial support of Drs. Sarah Wheelan and Vasan Yegnasubramanian at the Sidney Kimmel Comprehensive Cancer Center in the Department of Oncology.