MiXCR v4.4.0
🚀 New features
Built-in alleles database
MiXCR features robust support for inferring donor-specific allelic variants of V and J genes from NGS data, using the findAlleles command. With this new release, we introduce a comprehensive built-in database of human alleles. Now, the findAlleles command will utilize known allele names from this integrated library. Feel free to explore our database at https://vdj.online/library.
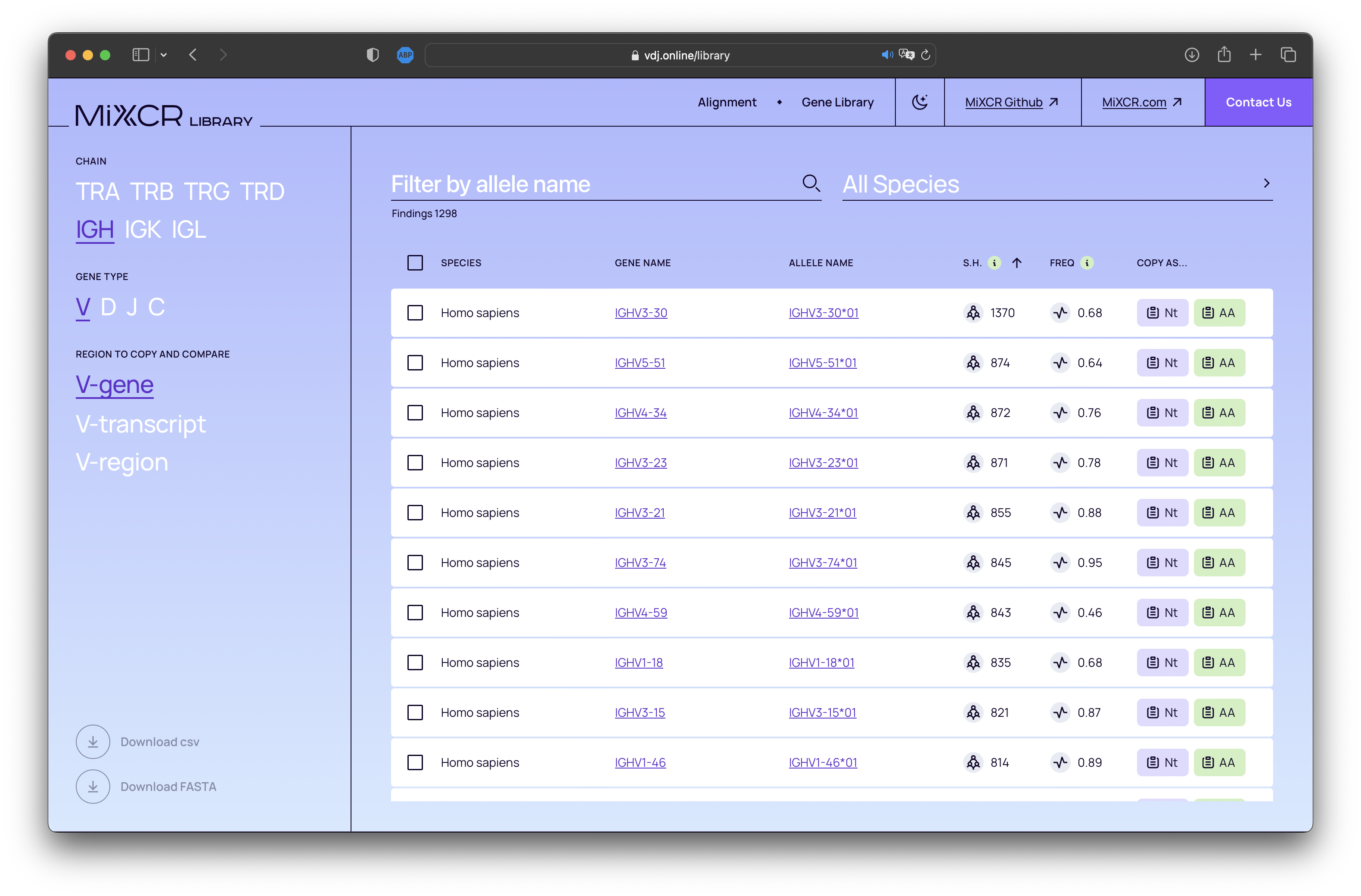
New rigorous quality checks
MiXCR now offers detailed insights into the quality of input data with its new quality control (QC) checks. A comprehensive list of checks provides complete information about the data and facilitates immediate feedback to the wet lab if any issues are detected
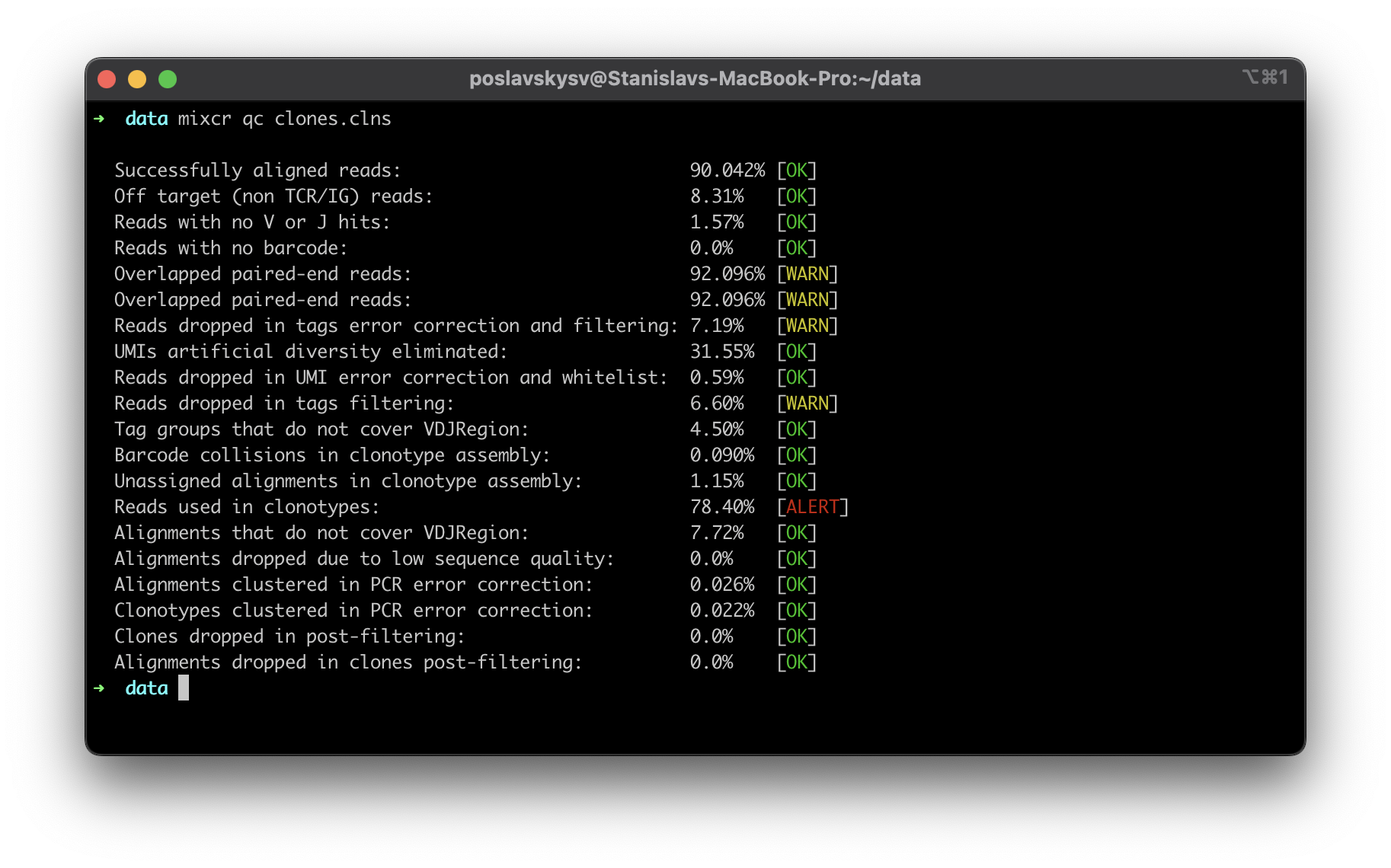
Convenient way to build custom libraries
Now one can build gene segment reference library for de-novo libraries or for chimeric model animals with just a single buildLibrary command. Check out our updated guide.
mixcr buildLibrary \
--v-genes-from-fasta v-genes.IGH.fasta \
--v-gene-feature VRegion \
--j-genes-from-fasta j-genes.IGH.fasta \
--d-genes-from-fasta d-genes.IGH.fasta \ # optional
--c-genes-from-fasta c-genes.IGH.fasta \ # optional
--chain IGH \
--species phocoena \
phocoena-IGH.json.gzComprehensive support of sample sheets
Now one can pass sample sheet directly to MiXCR analyze command as input. This way one can easily run MiXCR for arbitrary structure of input files, demultiplexed or not, with any type of multiplexing used:
mixcr analyze generic-sc-ht-vdj-amplicon --species hsa \
sample-sheet.csv \
output_prefix🤩 New presets
-
Support of MiLaboratories Human 7 Genes DNA Multiplex:
milab-human-dna-xcr-7genes-multiplex -
Support of Parse Bio Evercode Whole Transcriptome presets:
parsebio-sc-3gex-evercode-wt-mini,parsebio-sc-3gex-evercode-wtandparsebio-sc-3gex-evercode-wt-mega -
Support of FLAIRR-Seq protocol via
flairr-seqpreset -
New generic single cell presets:
-
Low throughput (e.g. micro-wells) amplicon-based single cell:
- No UMIs:
generic-sc-lt-vdj-amplicon - With UMIs:
generic-sc-lt-vdj-amplicon-umi
- No UMIs:
-
Low throughput (e.g. micro-wells) single cell with fragmentation (RNA-Seq):
- No UMIs:
generic-sc-lt-vdj-fragmented - With UMIs:
generic-sc-lt-vdj-fragmented-umi
- No UMIs:
-
High throughput (e.g. droplets) amplicon-based single cell:
- No UMIs:
generic-sc-ht-vdj-amplicon - With UMIs:
generic-sc-ht-vdj-amplicon-umi
- No UMIs:
-
High throughput (e.g. droplets) single cell with fragmentation (RNA-Seq):
- No UMIs:
generic-sc-ht-vdj-fragmented - With UMIs:
generic-sc-ht-vdj-fragmented-umi
- No UMIs:
-
Reconstructing VDJ from generic gene expression data:
- No UMIs:
generic-sc-gex - With UMIs:
generic-sc-gex-umi
- No UMIs:
-
-
New Biomed2 primer sets:
biomed2-human-rna-igkl,biomed2-human-rna-trbdg.
💪 Major changes
-
Improved aligner parameters for all protocols. We spent in total more than 100,000 CPU/hours running optimization. As a result alignment rate is better for most of the protocols, especially in the case of average data quality.
-
Adds new
minSequenceCountparameter for k-mer filter, allowing construction of more flexible filtering pipelines with better fallback behaviour for under-sequenced libraries. -
Now full sample sheet with input file names can be provided as an input to the pipeline.
-
Sample sheets provided both with
--sample-sheetmixin and as a pipeline input, will be fuzzy matched against the data, allowing for one substitutions in unambiguous cases. This behaviour can be turned off by using--sample-sheet-strictmixin instead, or by adding a--strict-sample-sheet-matchingoption if full sample sheet input is used as pipeline input. -
New commands:
mixcr qc,mixcr buildLibrary) ,mixcr mergeLibrary,mixcr debugLibrary) -
Various major improvements to sequencing and PCR error correction algorithms for tags and clonotypes:
- tag refinement now uses average quality in statistical inference; this is the correct approach from the mathematical point of view, and it slightly increases performance judging by better consensus assembly downstream
- statistical inference in PCR error correction redone from scratch, now it takes into account aggregated quality scores of clonotypes, which makes the procedure automatically adapt to low quality samples and better perform in many marginal cases in both UMI and non-UMI protocols
- better algorithm for quality score aggregation in clonotype assembly
- better algorithm for quality score aggregation in consensus assembly
-
Mechanism to apply different tag transformations on the
alignstep. Transformations include mappings, string and sequence manipulations and various arithmetic operations. This feature allows to fit single-cell scenarios where multiple well-known barcodes marks the same cell, allows to convert sequence barcodes to textual representation to adopt different barcode naming schemas used in some protocols, convert multiple barcodes to single cell id. Feature is currently used in presets for analysis of data from Parse Bioscience and BD Rhapsody single-cell platforms. -
Special mechanism to allow for
NaNvalues in metrics in group filters (used inminSequenceCountparameter in k-mer filter, see below). -
Added fallback behaviour for under-sequenced libraries
🐞 Bug fixes
- Fix for naming of intermediate files and reports produced by
analyzeif target folder is specified - Tag pattern now is also searched in reverse strand for single-ended input with
--tag-parse-unstranded - fix for value in report line
Reads dropped due to low quality, percent of total report string - Fixed bug not allowing to parse more than two reads with tag pattern
- Fixed bug when
--chainsis used withexportClonesOverlap - Fixed for
export...- tag quality field added back to export columns
👷 Minor fixes and improvements
- Added gene feature coverage in alignment report
- On Linux platforms default calculation of -Xmx now based on "available" memory (previously "free" was used)
- New gene aligner parameter
edgeRealignmentMinScoreOverridefor more sensitive alignments for short paired-end reads - Report values downstream
alignnow calculate percents relative to the number of reads in the sample rather than the
total number of reads in multi-sample analysis - Options helping with advanced analysis of data quality and consensus assembly process added
toassemble(--consensus-alignments,--consensus-state-stat,--downsample-consensus-state-stat)
andanalyze(--output-consensus-alignments,--output-consensus-state-stat,--downsample-consensus-state-stat) - Better tag pattern search projection representation in reports
findAllelesnow recalculate functionality of de novo found alleles- Better algorithm to calculating checksum of VDJC library
- Additional report string "Aligned reads processed" in
assemblereport - Added options
--by-featureand--by-genetosortClones - Added options
-rankByReadsand-rankByTag <(Molecule|Cell|Sample)>toexportClonesandexportShmTreesWithNodes.txt - Export
readIdsinexportAlignmentsby default - Added recalculation functionality for de-novo found alleles in
findAlleles - Add info about CDR3 in generated hash for de-novo alleles
- Remove de-novo alleles that are actually the same
findAlleleswill remove not used genes from the library (genes that not represented in given donor)- Make
--chainsoptional indownsamplingcommand and allow multiple input - Write empty file on
exportClonesif file doesn't contain any clones - Better exception messages on incorrect inputs for export commands
- In
exportCloneswriteno_d_geneif requestedVDJunction,DCDR3PartorDJJunctionin absence of D hit - Columns in
exportReportsTablenow covers most of significant statistics from reports
🐬 Docker image changes
-
Custom entry-point of the image removed, and now is set to
/bin/bash. Now one needs to specifymixcrcommand at the beginning of argument list:Old:
docker run ghcr.io/milaboratory/mixcr/mixcr analyze ...New:
docker run ghcr.io/milaboratory/mixcr/mixcr mixcr analyze ... -
New image is based on Amazon Corretto which in turn is based on Amazon Linux 2. If customization is required for the image, one now need to use
yumpackage manager instead ofapt/apt-get.With old image:
FROM ghcr.io/milaboratory/mixcr/mixcr:4.3.2 # ... RUN apt-get install -y wget # ...
With new image:
FROM ghcr.io/milaboratory/mixcr/mixcr:4.4.0 # ... RUN yum install -y wget # ...
see official docs for more detais.
-
Better compatibility of official docker image with Nextflow
‼️ Breaking changes
-
Parameter
clusteringFilter.specificMutationProbabilityremoved fromassembleaction. Three new parameters are introduced instead:-
clusteringFilter.correctionPower- this parameter determines how thorough the procedure should eliminate erroneous variants. Smaller value leaves less erroneous variants at the cost of accidentally correcting true variants. This value approximates the fraction of erroneous variants the algorithm will miss (type II errors). -
clusteringFilter.backgroundSubstitutionRate- expected rate of substitutions happening before the sequencing. -
clusteringFilter.backgroundIndelRate- expected rate of indels happening before the sequencing.
-
-
Majority of presets underwent name revisions (legacy names remain functional, though accompanied by deprecation warnings). See the full list of renames here.