![]()
![]()
msk/nucleovar is a bioinformatics pipeline that ...
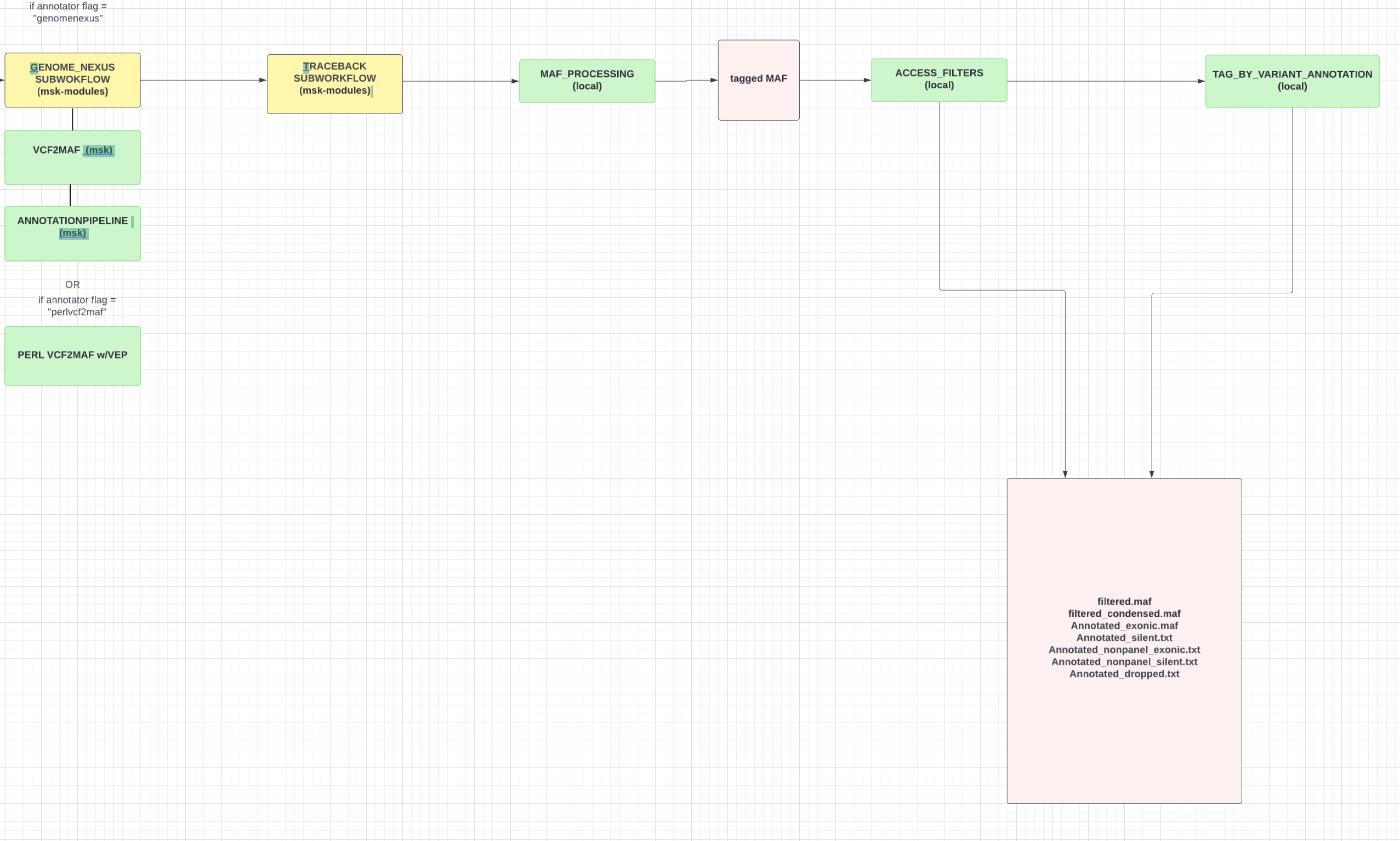
Processes a variety of sample BAM files through three variant callers (Mutect (v.1.1.5), VarDict, and GATK Mutect2). Output VCF Files are normalized, sorted, and concatenated, proceeding to be annotated and converted into a MAF format file. The following MAF file is tagged with the presence/absence of specific variant criteria, resulting in a final output MAF file containing variants filtered by criteria set forth by the ACCESS pipeline.
- Read in core samplesheet (containing case and control samples) and auxillary bams samplesheet
- Run case and control samples through variant callers (Mutect v.1.1.5, VarDict, and GATK Mutect2)
- Take output VCF Files from each variant caller and normalize, sort, concatenate and annotate using BCFtools suite.
- Convert output VCF File into MAF file format and annotate using Genome Nexus (option provided to invoke PERL VCF2MAF script as well)
- Tag output MAF file using MSK ACCESS pipeline criteria (presence of hotspots and removal of specific variant annotations)
- Run tagged MAF file through a traceback subworkflow which tags the file with presence of genotypes and performs specific tagging and concatentation.
- Tag output file is run through filtering based on criteria set forth by ACCESS_filters script.
Note
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
core_samplesheet.csv:
patient_id,sample_id,type,maf,duplex_bam,duplex_bai,simplex_bam,simplex_bai
PATIENT1,SAMPLE1,case,null,path/to/duplex.bam,path/to/duplex.bai,path/to/simplex.bam,path/to/simplex.bai
Each row represents an individual case and control sample.
aux_bams_samplesheet.csv:
sample_id,normal_path,duplex_path,simplex_path,type
SAMPLE1,/path/to/normal.bam,path/to/duplex.bam,path/to/simplex.bam,curated
Each row represents an individual sample which may contain a standard bam (if an unmatched or matched normal sample), or an individual sample which contains a simplex and duplex bam (if a curated or plasma sample)
Now, you can run the pipeline using:
nextflow run msk/nucleovar/main.nf \
--input core_samplesheet.csv \
--aux_bams aux_bams.csv \
--rules_json rules.json \
-profile <docker/singularity/.../institute> \
--outdir <OUTDIR>Warning
Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters;
see docs.
msk/nucleovar was originally written by @rnaidu and @buehlere.
We thank the following people for their extensive assistance in the development of this pipeline:
If you would like to contribute to this pipeline, please see the contributing guidelines.
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
This pipeline uses code and infrastructure developed and maintained by the nf-core community, reused here under the MIT license.
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.