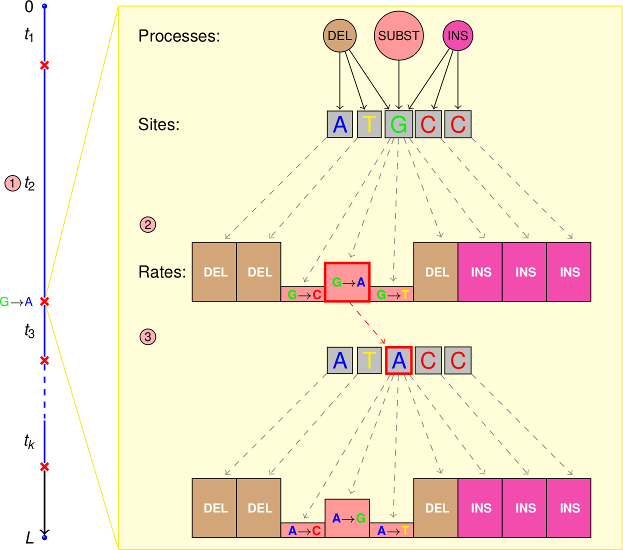
PhyloSim is an extensible object-oriented framework for the Monte Carlo simulation of sequence evolution written in 100 percent R. It is built on the top of the R.oo and ape packages and uses the Gillespie algorithm to simulate substitutions, insertions and deletions.
PhyloSim was brought to you by the Goldman group from EMBL-EBI.

Botond Sipos, Tim Massingham, Gregory E Jordan and Nick Goldman (2011) PhyloSim - Monte Carlo simulation of sequence evolution in the R statistical computing environment - BMC Bioinformatics 12:104 doi:10.1186/1471-2105-12-104
The released packages are available from CRAN.
-
Simulation of the evolution of a set of discrete characters with arbitrary states evolving by a continuous-time Markov process with an arbitrary rate matrix.
-
Explicit implementations of the most popular substitution models (nucleotide, amino acid and codon substitution models).
-
Simulation under the popular models of among-sites rate variation, like the gamma (+G) and invariant sites plus gamma (+I+G) models.
-
The possibility to simulate under arbitrarily complex patterns of among-sites rate variation by setting the site specific rates according to any R expression.
-
Simulation of one or more separate insertion and/or deletion processes acting on the sequences and which sample the insertion/deletion length from an arbitrary discrete distribution or an R expression (so all the probability distributions implemented in R are readily available for this purpose).
-
Simulation of the effects of variable functional constraints over the sites by site-process specific insertion and deletion tolerance parameters which determine the rejection probability of a proposed insertion/deletion.
-
The possibility of having a different set of processes and site-process specific parameters for every site, which allows for an arbitrary number of partitions in the simulated data.
-
The possibility to evolve sites by a combination of substitution processes along a single branch.
-
Simulation of heterotachy and other cases of non-homogeneous evolution by allowing the user to set "node hook" functions altering the site properties at internal nodes.
-
The possibility to export the counts of various events ("branch statistics") as phylo objects (see the man page of exportStatTree.PhyloSim).
-
See the man page of the PhyloSim class and the package vignette for more features and examples.
The package can be built from the source by issuing make pack on a *nix system. The building process need the standard unix tools, Perl and R with the ape, R.oo, ggplot2 and compoisson packages installed.