Documentation | Discussion Forum
We also have a slack channel for real-time discussion. If you want to join the channel, contact mufeili1996@gmail.com.
Deep learning on graphs has been an arising trend in the past few years. There are a lot of graphs in life science such as molecular graphs and biological networks, making it an import area for applying deep learning on graphs. DGL-LifeSci is a DGL-based package for various applications in life science with graph neural networks.
We provide various functionalities, including but not limited to methods for graph construction, featurization, and evaluation, model architectures, training scripts and pre-trained models.
For a list of community contributors, see here.
For a full list of work implemented in DGL-LifeSci, see here.
DGL-LifeSci should work on
- all Linux distributions no earlier than Ubuntu 16.04
- macOS X
- Windows 10
DGL-LifeSci requires python 3.6+, DGL 0.4.3+ and PyTorch 1.2.0+.
Additionally, we require RDKit 2018.09.3 for utils related to cheminformatics. We recommend installing it with
conda install -c rdkit rdkit==2018.09.3
For other installation recipes for RDKit, see the official documentation.
pip install dgllife
pip install hyperopt
conda install -c dglteam dgllife
If you want to try experimental features, you can install from source as follows:
git clone https://github.com/awslabs/dgl-lifesci.git
cd dgl-lifesci/python
python setup.py install
Once you have installed the package, you can verify the success of installation with
import dgllife
print(dgllife.__version__)
# 0.2.4If you are new to DGL, the first time you import dgl a message will pop up as below:
DGL does not detect a valid backend option. Which backend would you like to work with?
Backend choice (pytorch, mxnet or tensorflow):
and you need to enter pytorch.
To apply graph neural networks to molecules with DGL, we need to first construct DGLGraph --
the graph data structure in DGL and prepare initial node/edge features. Below gives an example of
constructing a bi-directed graph from a molecule and featurizing it with atom and bond features such
as atom type and bond type.
from dgllife.utils import smiles_to_bigraph, CanonicalAtomFeaturizer, CanonicalBondFeaturizer
# Node featurizer
node_featurizer = CanonicalAtomFeaturizer(atom_data_field='h')
# Edge featurizer
edge_featurizer = CanonicalBondFeaturizer(bond_data_field='h')
# SMILES (a string representation for molecule) for Penicillin
smiles = 'CC1(C(N2C(S1)C(C2=O)NC(=O)CC3=CC=CC=C3)C(=O)O)C'
g = smiles_to_bigraph(smiles=smiles,
node_featurizer=node_featurizer,
edge_featurizer=edge_featurizer)
print(g)
"""
DGLGraph(num_nodes=23, num_edges=50,
ndata_schemes={'h': Scheme(shape=(74,), dtype=torch.float32)}
edata_schemes={'h': Scheme(shape=(12,), dtype=torch.float32)})
"""We implement various models that users can import directly. Below gives an example of defining a GCN-based model
for molecular property prediction.
from dgllife.model import GCNPredictor
model = GCNPredictor(in_feats=1)For a full example of applying GCNPredictor, run the following command
python examples/property_prediction/classification.py -m GCN -d Tox21For more examples on molecular property prediction, generative models, protein-ligand binding affinity
prediction and reaction prediction, see examples.
We also provide pre-trained models for most examples, which can be used off-shelf without training from scratch.
Below gives an example of loading a pre-trained model for GCNPredictor on a molecular property prediction dataset.
from dgllife.data import Tox21
from dgllife.model import load_pretrained
from dgllife.utils import smiles_to_bigraph, CanonicalAtomFeaturizer
dataset = Tox21(smiles_to_bigraph, CanonicalAtomFeaturizer())
model = load_pretrained('GCN_Tox21') # Pretrained model loaded
model.eval()
smiles, g, label, mask = dataset[0]
feats = g.ndata.pop('h')
label_pred = model(g, feats)
print(smiles) # CCOc1ccc2nc(S(N)(=O)=O)sc2c1
print(label_pred[:, mask != 0]) # Mask non-existing labels
# tensor([[ 1.4190, -0.1820, 1.2974, 1.4416, 0.6914,
# 2.0957, 0.5919, 0.7715, 1.7273, 0.2070]])Similarly, we can load a pre-trained model for generating molecules. If possible, we recommend running the code block below with Jupyter notebook.
from dgllife.model import load_pretrained
model = load_pretrained('DGMG_ZINC_canonical')
model.eval()
smiles = []
for i in range(4):
smiles.append(model(rdkit_mol=True))
print(smiles)
# ['CC1CCC2C(CCC3C2C(NC2=CC(Cl)=CC=C2N)S3(=O)=O)O1',
# 'O=C1SC2N=CN=C(NC(SC3=CC=CC=N3)C1=CC=CO)C=2C1=CCCC1',
# 'CC1C=CC(=CC=1)C(=O)NN=C(C)C1=CC=CC2=CC=CC=C21',
# 'CCN(CC1=CC=CC=C1F)CC1CCCN(C)C1']If you are running the code block above in Jupyter notebook, you can also visualize the molecules generated with
from IPython.display import SVG
from rdkit import Chem
from rdkit.Chem import Draw
mols = [Chem.MolFromSmiles(s) for s in smiles]
SVG(Draw.MolsToGridImage(mols, molsPerRow=4, subImgSize=(180, 150), useSVG=True))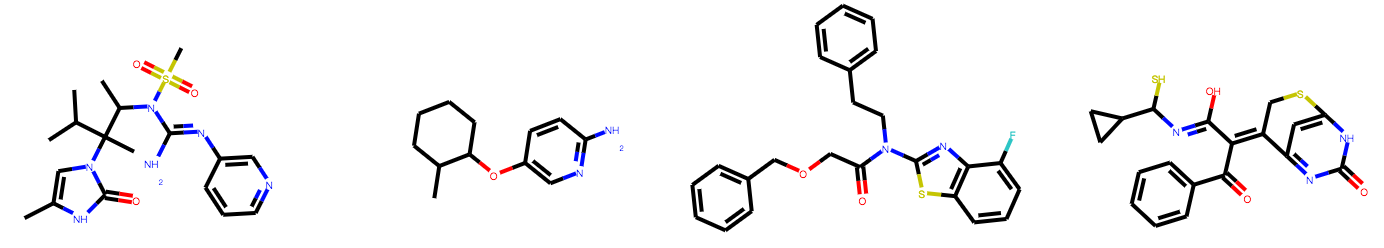
Below we provide some reference numbers to show how DGL improves the speed of training models per epoch in seconds.
| Model | Original Implementation | DGL Implementation | Improvement |
|---|---|---|---|
| GCN on Tox21 | 5.5 (DeepChem) | 1.0 | 5.5x |
| AttentiveFP on Aromaticity | 6.0 | 1.2 | 5x |
| JTNN on ZINC | 1826 | 743 | 2.5x |
| WLN for reaction center prediction | 11657 | 858 (1 GPU) / 134 (8 GPUs) | 13.6x (1GPU) / 87.0x (8GPUs) |
| WLN for candidate ranking | 40122 | 22268 | 1.8x |