Simplify GSEA and improve performance #119
Conversation
|
Check out this pull request on See visual diffs & provide feedback on Jupyter Notebooks. Powered by ReviewNB |
|
Thank you very much for your efforts @ivirshup ! |
|
View / edit / reply to this conversation on ReviewNB Zethson commented on 2022-11-29T15:58:14Z Nit: Guess I'd be happy if we could trim down some of the output here. ivirshup commented on 2022-11-30T12:51:37Z Fixed. That was left over from me trying to figure out how to move the pathways between R and python. Weirdly difficult! |
|
View / edit / reply to this conversation on ReviewNB Zethson commented on 2022-11-29T15:58:15Z Think this header is broken? It as a space before the ###? ivirshup commented on 2022-11-30T12:56:23Z It renders fine in the notebook view for me, and does not have a space there. Maybe it's a review-nb issue?
I also don't see this as a changed line with |
|
Fixed. That was left over from me trying to figure out how to move the pathways between R and python. Weirdly difficult! View entire conversation on ReviewNB |
|
It renders fine in the notebook view for me, and does not have a space there. Maybe it's a review-nb issue?
I also don't see this as a changed line with View entire conversation on ReviewNB |
|
@PauBadiaM I had some questions about switching some of the steps here to use decoupler. Different return valuesI'm getting different results from decoupler and the R packages (AUCell and fgsea). The results are qualitatively similar (e.g. similar rankings for pathways), but they are different. In particular I would say that the higher p-values from decoupler mainly pushed gene sets that don't really fit what we're looking at under any normal threshold. Any thoughts on this? Reactome pathways from omnipathI'm trying to decide on using the reactome pathways from the file downloaded in the tutorial, vs using omnipath. Right now I'm leaning towards using the file, since it's static along with the dataset. Any thoughts on this? Any plans to allow retrieving versions pathways from reactome? fryI don't believe |
|
Hi @ivirshup Nice! :) Regarding your questions:
Yes this is expected. AUCell resolves ties randomly, therefore results may change slightly. For
While this is nice for reproducibility, it is always good to be up to date to the latest version. In my opinion using a query wrapper is a better practice than manually downloading files, apart from having all the extra resources at hand instead of just one.
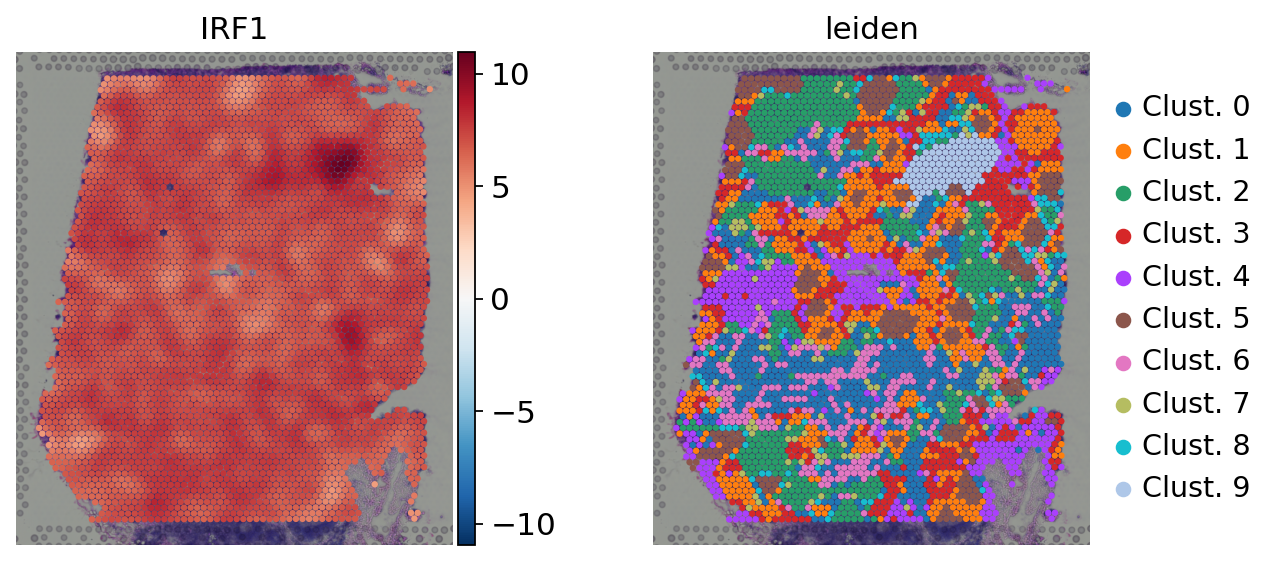
No it doesn't. The thing about Content wise, I would propose to make some additions/changes in this chapter: 1- Sample/cell vs contrast enrichment analysis: Make a discussion about that all enrichment tools can be used either at the sample/cell or at the contrast level. In the former, it helps to visualize and obtain gradients and variation of enrichment across your data. Example of TF enrichment in Visium slides, note the different spatial patterns at the spot level: In the later, its function is to summarize the obtained changes in expression after running a statistical DEG framework into interpretable terms. For example here are some DEGs that get summarized into pathway enrichments:
For the contrast part what I would do is to link to the 16. Differential gene expression analysis chapter, since the complex design of contrasts is already discussed there. 2- Distinction between gene sets and footprints: This chapter would benefit also by introducing the concept of footprint. The difference between a gene set and a footprint is that the former has no weights associated to the target genes, while the later does. This concept is crucial at estimating the direction of the enrichment. For example, let's say we estimate the enrichment of an inhibitory TF (with negative weights to its target genes). If done as a gene set (not considering the direction of the interactions), we might get a positive enrichment which is misleading. Happy to contribute writing sections/code! Also if needed we can meet online to discuss. |



I would agree using a query is better practice generally. My main concern here was using a fixed dataset, while the resource may update genome annotation names. Ideally I would be able to access older versions of reactome annotations, or at least have a good way of resolving possible annotation changes. What I think we should go with here is showing how one would query for the resource (in an unexecuted cell), but read from the file for stability of the tutorial. Re: content additions, those sound good, but I would defer to the editors (@Zethson, @soroorh, @alitinet) on this. My initial interest here was just updating the sampling function, but then I couldn't import AUCell, then had to rewrite some bits so did a broader cleanup. So I'd frame this PR as more of an editing pass than a content update. I'd suggest we quickly finish up this PR, then make a content PR separately? |
This seems like a good option.
Sounds good! Just one minor thing before merging, if possible please add For the content additions let's see what the editors think. |
I just read your excellent description of your content suggestions and I'll keep it brief: Yes to all - happy to review and accept a PR :) |
|
I think this is ready for a review. Please let me know if there's anything else to change. One last big change I made: since all the gene set enrichment plots were basically the same, I just did them all the same way with |
|
Looks good! Just one small comment, decoupler does filter by gene set size with the |
View entire conversation on ReviewNB |
|
I had no idea. Thanks! Then keep it as it is of course :) View entire conversation on ReviewNB |
|
I think people are going to skip around the notebooks. I would also note that in the previous version the first two commands were done "wrong" – so it might be worth documenting the intent of the commands. The counts layer was a reference to View entire conversation on ReviewNB |
|
All right. I'm not going to veto this so feel free to keep it View entire conversation on ReviewNB |
|
As with the comment above:
I think each block of code should have some documentation about what it's meant to do. This is how the rest of the notebook is written.
We could swap some of these comments with a cell that says "this code is doing the preprocessing"? View entire conversation on ReviewNB |
|
Nah, I'd rather have the explicit comments that you currently have than such a cell. View entire conversation on ReviewNB |
|
Pretty much.
I could just do the View entire conversation on ReviewNB |
|
Please do. No need to import Union though then, right?
View entire conversation on ReviewNB |
Sure. Any suggestions for how to do this other than just filtering the warning would be appreciated though! |
|
View / edit / reply to this conversation on ReviewNB soroorh commented on 2022-12-09T12:14:36Z Thanks for the efforts. I'm not really happy with the pseudo-bulking approach used here, I'm afraid, specifically with the idea of sampling a fixed number of cells per cell type. Note that by this step, you've already performed library depth normalisation (which i compared against
A few of things are well known about this dataset -
and none of these are captured in the proposed pseudo-bulk approach here
Given that you've already imported decoupler, i suggest we go with their pb approach as follows:
# randomly assign cells to replicates
adata.obs["pseudoreplicate"] = np.random.choice(["R1", "R2", "R3"], size=adata.n_obs)
adata.obs['SampleID'] = adata.obs.pseudoreplicate.astype('str') + '_' + adata.obs.label.astype('str')
This gives results very similar to original plots and consistent to biology (note I am assuming adata is not subsetted on HVGs, as you've done here)
This is what you expect, in case it helps AnnData object with n_obs × n_vars = 42 × 15361
|
|
View / edit / reply to this conversation on ReviewNB soroorh commented on 2022-12-09T12:20:03Z You've inconsistent reference to ivirshup commented on 2022-12-11T19:23:49Z I've updated the text. Let me know if the changes are alright. soroorh commented on 2022-12-16T06:43:40Z looks good - thanks |
|
View / edit / reply to this conversation on ReviewNB soroorh commented on 2022-12-09T12:23:00Z Thanks a heap for adding this function. I also think we should go with static file. A part from reproducibility issues, i realised that not all MsigDB collection (e.g., GO terms) can be queried by |
|
View / edit / reply to this conversation on ReviewNB soroorh commented on 2022-12-09T12:41:49Z Please check this when you tried pb with ivirshup commented on 2022-12-11T19:16:54Z As noted above, this seems like biological signal rather than evidence of noise. |
|
View / edit / reply to this conversation on ReviewNB soroorh commented on 2022-12-09T13:26:05Z These results are way too different from the original results - these suggest a strong MAPK signalling, but since PC1 is , as of now, confounded with lib size, it is very difficult to say if MAPk signaling hints are related to biology or technical effects. Hopefully we get a better picture in re-run. ivirshup commented on 2022-12-11T18:39:06Z The FDRs for this comparison start well below the lowest FDR we see from the previous contrast. Given the lack of interpretation of these results, I read this as saying "there is no significant enrichment".
I think the distribution of FDRs supports this:
with plt.rc_context({"figure.figsize": (10, 5)}):
sns.histplot(
(
pd.concat(
{
"first_comparison": fry_results,
"second_comparison": fry_results_negative_ctrl
},
names=["comparison"]
)
.assign(**{"-log10(FDR)": lambda x: -np.log10(x["FDR"])})
),
x="-log10(FDR)",
hue="comparison"
)
soroorh commented on 2022-12-16T06:56:37Z The point of this section was just to demonstrate how to deal with complicated contrasts. Lack of good bio interpretation for the specific contrast chosen here was confusing - so in the revised version this will be replaced with a more biologically meaningful contrast with known ground truth. |
|
View / edit / reply to this conversation on ReviewNB soroorh commented on 2022-12-09T13:29:48Z the least ranked genes for me are %%R tail(ranked_genes) [1] "CCR7" "LAT" "CD7" "ITM2A" "OCIAD2" "CD3D" So, the ranks will change. This is a dataset where the perturbation effect is so strong and therefore ranking won't change substantially in subsetted data vs full data. I'll have to have a thing about this step.
ivirshup commented on 2022-12-11T19:15:43Z Note that I sorted by *absolute value* instead of strictly by score. The genes mentioned have large negative scores. |
|
ReviewNB is reminding me why I try not to use it, so I'm just gonna respond here.
The FDRs for this comparison start well below the lowest FDR we see from the previous contrast. Given the lack of interpretation of these results, I read this as saying "there is no significant enrichment". I think the distribution of the FDRs suggests this is the right approach: with plt.rc_context({"figure.figsize": (10, 5)}):
sns.histplot(
(
pd.concat(
{
"first_comparison": fry_results,
"second_comparison": fry_results_negative_ctrl
},
names=["comparison"]
)
.assign(**{"-log10(FDR)": lambda x: -np.log10(x["FDR"])})
),
x="-log10(FDR)",
hue="comparison"
)
|

I don't think this is clear to me. PC1 shows a strong separation between monocytic and lymphatic lineage. While library size is confounded by lineage I would note that there isn't a monotonic relationship between PC1 and library size. I can add a note explaining this, but could also just not bother with plotting library size here.
I was a little confused by this comment. To my mind the plots show all of these things, but I'll respond bullet by bullet.
They group by condition, and by cell type along the first PC.
The second PC separates all cell types by stimulation
While I think it's very possible there are effects in other PCs, I think we do see that effect in my version. Calculating the mean distance in the first two PCs by cell type: obsdf = sc.get.obs_df(pb_data, ["cell_type", "condition"], [("X_pca", 0), ("X_pca", 1)])
obsdf.groupby(["cell_type", "condition"]).mean().groupby("cell_type").diff()The absolute difference between the monocytes is higher in the second PC. Finally, about whether one should use a fixed number of cells per sample. My argument would be that: we are trying to look at the differences between cell types, not cell type proportions. So, if there would be some effect due to cell type proportion, we should try to mitigate that. If we subsample to a single cell type, and sample different numbers of cells, we can see an effect. I will subsample to CD4 T cells and show this cd4_t_cells = adata[adata.obs["cell_type"] == "CD4 T cells"].copy()
adatas = {}
cell_counts = [75, 150, 300, 600, 1200]
for n_cells in cell_counts:
adatas[str(n_cells)] = subsampled_summation(cd4_t_cells, ["cell_type", "condition"], layer="counts", n_samples_per_group=3, n_cells=n_cells)
pb_tcells = ad.concat(adatas, label="n_cells", index_unique="-")
pb_tcells.obs["n_cells"] = (
pb_tcells.obs["n_cells"]
.astype(int)
.astype(pd.CategoricalDtype(categories=cell_counts, ordered=True))
)
pb_tcells.obs["lib_size"] = pb_tcells.X.sum(axis=1)
pb_tcells.layers["counts"] = pb_tcells.X.copy()
sc.pp.normalize_total(pb_tcells)
sc.pp.log1p(pb_tcells)
sc.pp.pca(pb_tcells)
sc.pl.pca(pb_tcells, color=["n_cells", "lib_size", "condition", "cell_type"], size=250)
I did try the suggested approach, and it gives less useful PCs, notably making the treatment effect no longer orthogonal to cell type. Code to computeadata_pseudo = adata.copy()
adata_pseudo.obs["pseudoreplicate"] = np.random.choice(["R1", "R2", "R3"], size=adata_pseudo.n_obs)
adata_pseudo.obs['SampleID'] = adata_pseudo.obs.pseudoreplicate.astype('str') + '_' + adata_pseudo.obs.condition.astype('str')
pb_unbalanced = decoupler.get_pseudobulk(adata_pseudo, sample_col='SampleID', groups_col='cell_type', layer='counts', min_cells=75, min_prop=0).copy()
pb_unbalanced.obs['lib_size'] = pb_unbalanced.X.sum(1)
# Normalize
sc.pp.normalize_total(pb_unbalanced)
sc.pp.log1p(pb_unbalanced)
# compute PCA and plot
sc.pp.pca(pb_unbalanced)# Using decoupler style pseudo bulking
sc.pl.pca(pb_unbalanced, color=["condition", "lib_size", "cell_type"], size=250)
# Accounting for dataset composition (e.g. how I approached this)
sc.pl.pca(pb_adata, color=["condition", "lib_size", "cell_type"], size=250)
|



|
Note that I sorted by *absolute value* instead of strictly by score. The genes mentioned have large negative scores. View entire conversation on ReviewNB |
|
As noted above, this seems like biological signal rather than evidence of noise. View entire conversation on ReviewNB |
|
I've updated the text. Let me know if the changes are alright. View entire conversation on ReviewNB |
|
Added. View entire conversation on ReviewNB |
|
looks good - thanks View entire conversation on ReviewNB |
|
The point of this section was just to demonstrate how to deal with complicated contrasts. Lack of good bio interpretation for the specific contrast chosen here was confusing - so in the revised version this will be replaced with a more biologically meaningful contrast with known ground truth. View entire conversation on ReviewNB |
|
Given the lack of proper evidence (publications) on whether it is correct to pseudo-bulk a fixed number of cells per cell type and the whole confusions around potential confounding with cell type proportion, the pseudo-replication based workflow is going to be replaced with standard sample-based workflow, addressed in another issue. |
|
Thanks all involved in the issue. Further update plans for this chapter are described in #132 |
Ping @Zethson @soroorh
Simplified GSEA notebook + performance improvements.
Simplifications include:
Performance improvements include:
XsparseTODO:
If so, removeedgerdependencydecoupler's API (maybe not on binder, but I think the whole thing is a problem on binder)