Home
The scCancer package focuses on processing and analyzing droplet-based scRNA-seq data for cancer research. Except basic data processing steps, this package takes several special considerations for cancer-specific features.
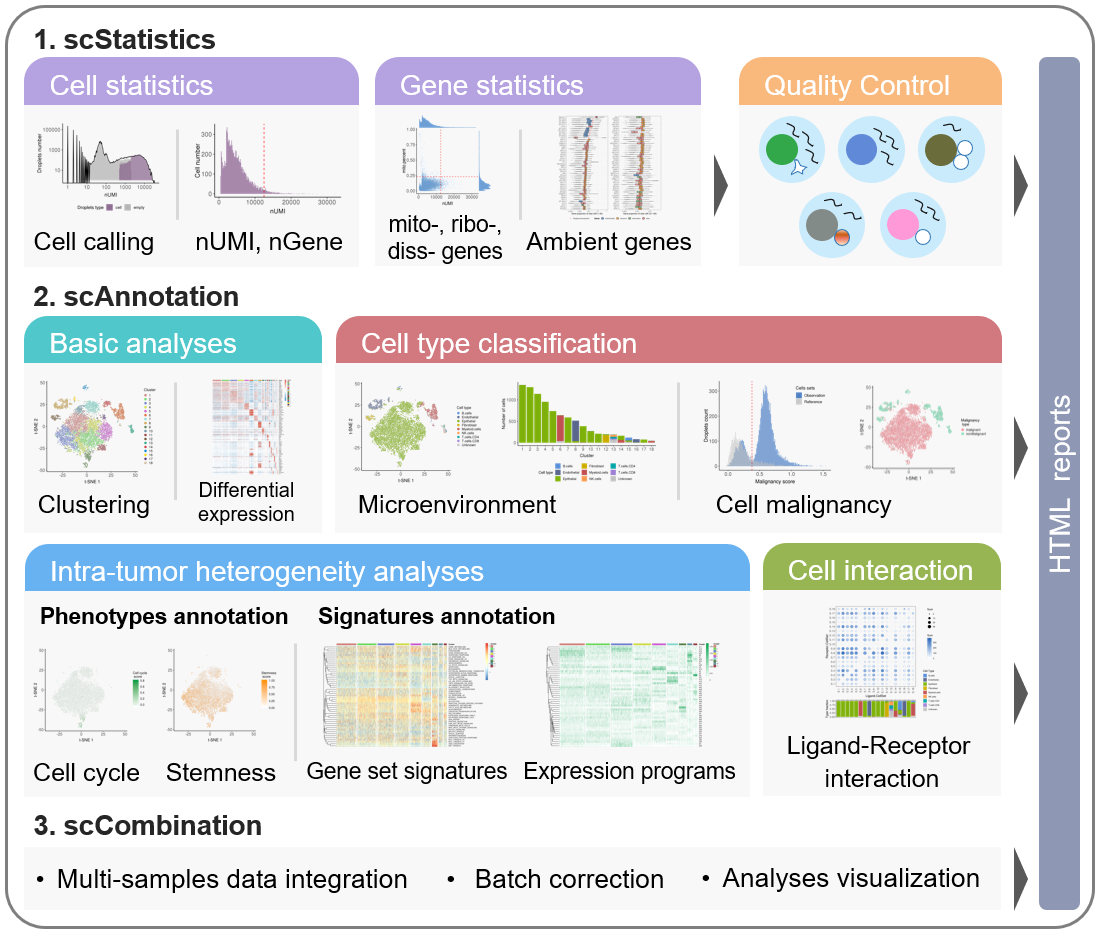
The workflow of scCancer mainly consists of two parts: scStatistics and scAnnotation.
- The
scStatisticsperforms basic statistical analysis of raw data and quality control. - The
scAnnotationperforms functional data analyses and visualizations, such as low dimensional representation, clustering, cell type classification, malignancy estimation, cellular phenotype scoring, gene signature analysis, etc.
After these analyses, user-friendly graphic reports will be generated.
- Memery: >= 32G (for a data with ~10000 cells)
- R version: >= 3.5.0
Firstly, please install or update the package devtools by running
install.packages("devtools")Then the scCancer can be installed via
library(devtools)
devtools::install_github("wguo-research/scCancer")The vignette of scCancer can be found in the project wiki or vignette page.
We also provide an example data of kidney cancer from 10X Genomics, and here are the generated reports:
Please use the following citation:
GPL-3